
Better Immunodetection
Information on antibodies, their selection, and use.
On This Page | Detection Overview | Blocking | Primary Antibodies | Secondary Antibodies | Detection Methods | Total Protein Detection | Tips & Protocols |
Sign Up for Western Blotting Tips and Webinar Alerts
Detection Overview
After transfer, proteins from the gel will be immobilized on the membrane. This section describes the process of specifically detecting your particular protein of interest in order to identify and quantify its presence and abundance in the sample. Western blot detection of proteins utilizes primary antibodies that are specific for the target protein which are then in turn recognized by secondary antibodies that are conjugated with enzymes or fluorescent molecules for detection. The correct selection and use of appropriate primary and secondary antibodies is crucial for maximizing signal while reducing background. The proper use of protein loading controls are also critical for signal normalization in quantitative western blotting. We discuss the most popular methods to detect total protein on the membrane for loading controls and their relative advantages and disadvantages.
The Immunodetection Workflow

Western Blotting Antibody Detection
After proteins are transferred from the gel to the membrane, antibodies specific to your protein of interest (primary antibodies) are incubated with the membrane to allow them to recognize their targets. A second incubation with conjugated antibodies specific to the primary antibodies (secondary antibodies) enables visualization by various methods.
Block unbound membrane sites
Membranes are incubated in a blocking agent that masks any sites on the membrane that would allow antibodies to bind non-specifically. Failure to do so raises background.
Incubate with primary antibody. Wash away excess.
Membranes are then incubated with primary antibodies that are specific to your protein of interest. These bind to their targets and any excess is washed away.

Incubate with secondary antibody. Wash away excess.
The membrane is then incubated with secondary antibodies that recognize the primary antibodies and are also conjugated to either an enzyme or a fluoroscent molecule. Any excess is washed away.

Develop signal
For secondary antibodies conjugated with an enzyme, the membrane is incubated with a chemical substrate that produces a signal that can then be detected. If the secondary is conjugated to a fluorescent label, the membrane can be directly imaged in an instrument capable of fluorescent imaging.
Blocking
Following transfer, unoccupied antibody binding sites on the membranes must be blocked to prevent nonspecific binding. Failure to completely block these sites can lead to high backgrounds that obscure the signal.
Various blocking reagents are available, and no single blocker will be optimal for all antibody/antigen combinations. The best blocker for each experiment will depend on the antibody and membrane type. Optimize the detection system for maximal signal with lowest background by testing several blocking agents. Modern formulations like Bio-Rad’s EveryBlot Blocking Buffer are universal, performing well across a wide range of targets, sample types, and detection methods.
Blocking duration also affects the signal and background levels. Blocking for 1 hour with constant agitation is a good starting point. Excessive blocking times may result in lower sensitivity as epitopes can be masked by the blocking agent and proteins washed off the membrane. In contrast, insufficient incubation times will increase nonspecific binding of the primary antibody. Advanced formulations like Bio-Rad’s EveryBlot Blocking Buffer can complete the blocking step in 5 minutes.
For detection of a phosphorylated form of a protein, blocking buffers should not contain phosphorylated proteins to avoid high background signal. Avoid use of nonfat dry milk and instead use BSA, casein, or advanced formulations that are compatible with phosphoprotein detection.
Quick Tips:
Optimizing the Blocking Step in Western Blotting
Watch the video to understand the experimental parameters and which type of blocking agent to use to ensure success of your western blot.
| Agent | Pros | Cons |
|---|---|---|
| Non-fat milk (1–5%) | Inexpensive, widely available | Not ideal for phosphoproteins, biotin-avidin/streptavidin, or AP detection Speckling if not completely solubilized |
| BSA (0.5–5%) | Recommended for phosphoprotein detection, AP/biotin-avidin detection | Not usable with antibodies created using BSA-coupled peptides, phosphotyrosine Ab |
| Casein (1–2%) | Balance of stringency & sensitivity Ideal for most detection methods | Blocking may be insufficient for complex biological samples |
| Gelatin (1–5%) | Inexpensive, widely available Fish gelatin does not cross-react with mammalian proteins | Porcine gelatin solidifies in cold blocking Speckling if not completely solubilized Cannot be used with avidin-biotin detection |
| Specialized | Advantage depends on product | Can be expensive |
Primary Antibodies
An antibody is an immunoglobulin protein such as lgG that is generated in response to exposure to a foreign substance, or antigen. Antibodies have specific affinity for the antigens that elicited their synthesis.
Primary Antibodies

Primary antibodies are raised against a protein of interest and will selectively recognize and bind to target proteins that have been immobilized to a membrane. Antibodies can be designed to be specific for certain parts of a target protein, or they can be designed to be specific for only modified versions of the protein.
Primary Antibody Selection
Antibodies should be specific, selective, and give reproducible results. To ensure the specificity of the primary antibody, it is important to use positive and negative controls when running your blot. As a positive control, purified proteins or lysates overexpressing the target protein can be used. As a negative control, you can include secondary-antibody-only controls (omitting the primary antibody incubation step) and samples from a tissue or cell lysates known not to express the target protein such as cells engineered with a genetic knockout. Before performing the experiments review the literature in order to understand the expression profile of the protein
Selecting Primary Antibodies for Best Results in Western Blotting
Choosing a Primary Antibody for Multiplex Western Blotting

How to Select the Right Antibody
Bio-Rad has developed tips to consider when choosing your antibody.
The wording good and bad antibody or the most specific antibody should be avoided, since a specific antibody in one sample context can give rise to high cross-reactivity in another sample context depending on the nature of the epitope(s) that it will recognize.
When selecting a phospho-specific antibody for your experiments, it is crucial to ensure that the antibody specifically detects the protein of interest only when it is phosphorylated at the indicated site. You might also want to consider treating cells with growth factors or chemical compounds that induce or inhibit expression of the target.
Monoclonal vs. Polyclonal Antibodies
The antibodies used to detect the target protein in a western blot will be either monoclonal or polyclonal. Polyclonal antibodies consist of a mixed pool of immunoglobulin molecules that bind to several different epitopes found on a single antigen. Polyclonals are usually produced in rabbits, donkeys, sheep, and goats, and are purified from serum.
In contrast, monoclonal antibodies bind to a single epitope within a target antigen. They are composed of homogeneous cloned immunoglobulin molecules, rather than the heterogeneous antibody mixture typical of polyclonals. Monoclonals are made by fusing antibody-producing cells from the spleen of the immunized animal (usually a rat or mouse) with an immortalized cell line to produce single specificity antibodies that can be purified from tissue culture supernatant.
| 
| |
|---|---|---|
| Specificity | Specificity for a single epitope. | Varying specificities to multiple epitopes |
| Identification | Identifies whether a particular region of a protein is present | Identifies the entire target protein via binding at multiple sites. Since multiple epitopes are targeted, there is a higher likelihood of detection of the target |
| Cross-Reactivity | May cross-react with other proteins that share this epitope, such as isomers or common motifs | Higher background and cross-reactivity possible due to detection of multiple epitopes, any of which may be shared by related proteins |
| Sensitivity | Usually less sensitive since only a single antibody molecule binds to each target | More sensitive because signal is amplified through the binding of several antibodies per target |
| Cost | More expensive to produce initially, but available in an unlimited supply | Less expensive to produce initially, but supply is limited to immunized animal(s). Greater variability between preparations |
Primary Antibody Incubation
After blocking, the membrane is incubated in a solution containing the primary antibody, usually diluted in blocking buffer. The time and temperature of incubation depends on the binding affinity of the antibody to the target protein and should be determined for each antibody individually. One hour at room temperature with gentle agitation is a good starting point. In order to reduce the background staining, the amount of Tween 20 used in the buffers is also important.
Antibody Concentration
The optimum antibody concentration is the dilution of antibody that still yields a strong positive signal without background or nonspecific reactions. Instructions for antibodies obtained from a manufacturer typically suggest a starting dilution range. For custom antibodies or for those where a dilution range is not suggested, good starting points are:
- 1:100–1:1,000 dilution when serum or tissue culture supernatants are the source of the primary antibody
- 1:500–1:10,000 dilution of chromatographically purified monospecific antibodies
- 1:1,000–1:100,000 dilution may be used when ascites fluid is the source of antibody
Quick Tips:
How to Optimize Primary Antibody Concentration and Incubation for Western Blots
Watch this video to explore considerations for optimizing incubation time and dilution of primary antibodies when performing a western blot.
See Our Western Blotting Primary Antibodies
Phospho-Specific Antibodies
Protein phosphorylation, the addition of a phosphate group to serine, threonine or tyrosine residues, is an important cellular process utilized to send cellular signals from the membrane to the nucleus. Protein phosphorylation may result in conformational changes that trigger the activation or inactivation of an enzyme.
Phospho-specific antibodies are primary antibodies that detect only the phosphorylated forms of proteins and recognize the phosphorylated serine, threonine, or tyrosine residues in the context of the rest of the protein. Using a combination of total protein detection (using primary antibodies that recognize both phosphorylated and non-phosphorylated forms of the protein) and phospho-specific antibodies, it is possible to assess the degree of phosphorylation for any given protein.
Prior to using phospho-specific antibodies, ensure that the antibodies have been validated for your experimental system. This can be accomplished by using positive and negative controls. For example, lysates from cells that are either untreated or have been treated to stimulate the pathway of interest can act as negative and positive controls. Likewise, absence of detection can act as a negative control on lysates that have been treated with lambda phosphatase to remove phosphate groups.
Secondary Antibodies
Purification of Cross-Adsorbed Antibodies

A solution of secondary antibodies raised against mouse lgG is passed over a column containing immobilized serum proteins from potentially cross-reactive species such as rat or rabbit.
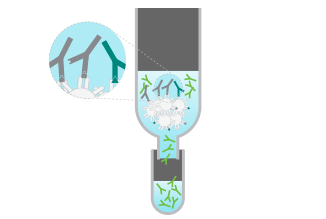
Only antibodies highly specific for mouse lgG will flow through the column, while antibodies cross-reacting to rat or rabbit will bind and remain in the column. The flow-through solution contains antibodies that specifically recognize mouse lgG.
Secondary antibodies are specific for the isotype and species of the primary antibody. For example, a goat anti-rabbit secondary is an antibody raised in goats against a primary antibody raised in rabbits.
Secondary antibodies bind to a number of different conserved regions on the primary antibody, and act to amplify the signal, increasing detection sensitivity. Secondary antibodies are labelled with either an enzyme for colorimetric or chemiluminescent detection or with a fluorescent dye for fluorescent detection of the protein of interest.
Cross-Adsorbed Antibodies
Secondary antibodies raised against primary antibodies of one species can recognize those from other species, especially if they are from closely related animals. For example, parts of the constant regions of mouse and rat antibodies are evolutionarily conserved, leading to conserved epitopes between both species. Since secondary antibodies are generally polyclonal, when generating secondaries against mouse, a subset of those secondaries will be specific for those conserved epitopes, being able to recognize them whether the epitope resides on a mouse or rat antibody. This cross-reactivity could lead to unexpected bands when multiplexing or cause high background.
To remove the undesired antibodies from a polyclonal pool, an affinity chromatography column is used to purify the secondary antibody preparation of offending cross-reactive species. Clonal antibodies that recognize the immobilized antigen(s) are removed. The unbound pool of antibodies is now cross-adsorbed against the immobilized antigen and should show no reactivity towards it.
The most common type of cross-adsorbed secondary antibody is species specific, which is most useful for multiplexing, although there are also instances when isotype-specific antibodies are required. For example, immunization with a purified IgG1 preparation might be expected to generate a serum with a specific reactivity towards IgG1 and no cross-reactivity with other antibody isotypes. However, due to the polyclonal nature of the serum combined with the conservation of some epitopes between classes and isotypes, a component of the polyclonal serum may react with an epitope on IgG1 that is also found on IgG2a, confounding the isotype specificity.
A solution of secondary antibodies raised against mouse IgG is passed over a column containing immobilized serum proteins from potentially cross-reactive species such as rat or rabbit.
Only antibodies highly specific for mouse IgG will flow through the column, while antibodies cross-reacting to rat or rabbit will bind and remain in the column. The flow-through solution contains antibodies that specifically recognize mouse IgG.
Secondary Antibody Concentration
Similar to primary antibodies, the optimum antibody concentration is the dilution of antibody that still yields a strong positive signal without background or nonspecific reactions. Fortunately, high-quality secondary antibodies are commonly available, and manufacturers typically suggest a starting dilution range.
See Our Western Blotting Secondary Antibodies
Quick Tips: How to Choose Secondary Antibodies for Multiplex Western Blotting
In this video, we discuss the best practices for selecting secondary antibodies that are compatible with your primary antibodies and your detection methods to produce high-quality, reproducible results.
Detection Methods
In the past, many different methods were used for western blot detection, but now the vast majority employ enzymatic chemiluminescence or fluorescent detection. Thus, most secondary antibodies are conjugated to an enzyme such as alkaline phosphatase (AP) or horseradish peroxidase (HRP) for use with a chemiluminescent substrate or labeled with a fluorescent compound for imaging.
Chemiluminescence Detection
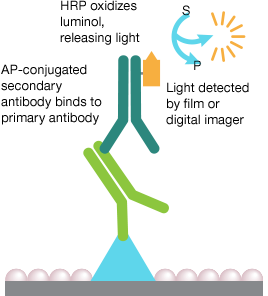
Chemiluminescence is a chemical reaction in which the oxidation of a chemical substrate such as luminol is catalyzed by an enzyme, typically horseradish peroxidase (HRP), and the reaction produces light as a byproduct. The resulting light can be captured on film or by a digital imager.
Luminol emits light only weakly, so enhancers are added to the reaction to increase the signal. This enhancement of a luminol-based signal is commonly referred to as enhanced chemiluminescence (ECL). There are a number of different enhancers available, some of which can increase the signal by as much as a 1,000-fold, making ECL more sensitive than other common detection systems such as conversion of substrates to colored precipitates.
The light intensity will be roughly proportional to the quantity of your protein of interest, allowing semi-quantitation of relative protein abundance. Since this detection method relies on an enzyme-substrate interaction, the kinetics of the reaction plays a role in the linearity of the signal. Therefore, for more accurate quantitation, care must be taken to ensure that the sample load is within the linear range of the assay and the detection signal is not saturated.
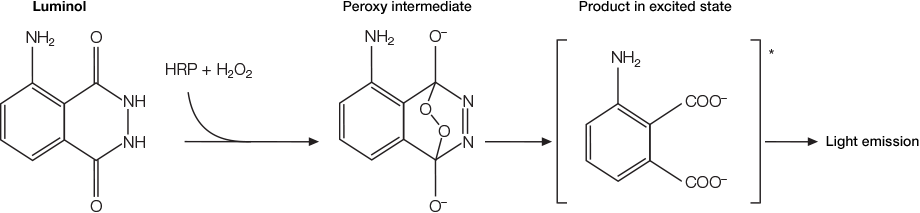
Luminol oxidized by HRP in the presence of H2O2 leads to the formation of a 3-aminophthalate dianion and the release of light.
Quick Tips:
How to Image a Chemiluminescent Western Blot
Watch this video to see how to prepare a western blot membrane for chemiluminescent detection and ensure a strong chemifluorescent signal during image acquisition.
See Our Chemiluminescent Substrates »
Fluorescence Detection

In fluorescence detection, a primary or secondary antibody is labeled with a fluorescent molecule (a dye or fluorophore). A light source that produces photons within the fluorophore's excitation spectrum excites the fluorophore, and the fluorophore then emits photons with a longer wavelength. The difference in wavelength between excitation and emitted light is termed Stokes shift. This emitted light of longer wavelength can then be distinguished from the excitation light via appropriately designed optical filters and detected by a digital imager.
Fluorescence detection has a number of advantages over traditional chemiluminescent detection. As fluorescent detection does not rely on an enzyme-substrate reaction, the use of fluorophores can be more quantitative than chemiluminescent detection, is often faster, and reduces waste.
The greatest advantage of fluorescent western blotting detection over chemiluminescent detection is the ability to simultaneously detect a large number of proteins on one blot, using a process called multiplexing. This relies on choosing fluorophores that fluoresce at different wavelengths and can be distinguished by the digital imager.
Fluorophore Selection
When using fluorescence detection, consider the following optical characteristics of the fluorophores to optimize the signal:
- Quantum yield — efficiency of photon emission after absorption of a photon. Processes that return the fluorophore to the ground state but do not result in the emission of a fluorescence photon lower the quantum yield. Fluorophores with higher quantum yields are generally brighter.
- Extinction coefficient — measurement of how well a fluorophore absorbs light at a specific wavelength. Since absorbance depends on path length and concentration (Beer's Law), the extinction coefficient is usually expressed in cm -1 M-1. As with quantum yield, fluorophores with higher extinction coefficients are usually brighter.
- Stokes shift — the difference in the maximum excitation and emission wavelengths of a fluorophore. Since some energy is dissipated while the fluorophore is in the excited state, emitted photons are of lower energy (longer wavelength) than the light used for excitation. Larger Stokes shifts minimize overlap between the excitation and emission wavelengths, increasing the detected signal.
- Excitation and emission spectra — excitation spectra are plots of the fluorescence intensity of a fluorophore over the range of excitation wavelengths; emission spectra show the emission wavelengths of the fluorescing molecule. Choose fluorophores that can be excited by the light source in the imager and that have emission spectra that can be captured by the instrument. When performing multiplex western blots, choose fluorophores with widely separated emission spectra to enable good signal separation among the different color channels.
Fluorophore Stokes Shift
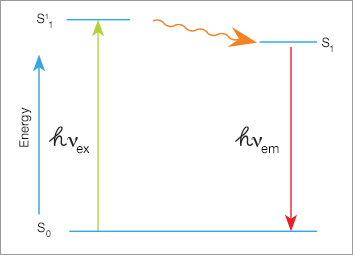
A high-energy photon excites a fluorophore, causing it to leave the ground state (S0) and enter a higher energy state (S11). Some of this energy dissipates, allowing the fluorophore to enter a relaxed excited state (S1). When the fluorophore returns to the ground state, a photon of light is emitted. Since some of the energy was dissipated, the emitted photon is of lower energy (longer wavelength).

The difference in wavelength between the exciting light and the emitted light is called the Stokes shift. Larger Stokes shifts minimize the overlap between the excitation and emission wavelengths, increasing the detection signal-to-noise ratio.
See Our Products for Fluorescent Western Blotting Detection »
Total Protein Detection
Total protein staining provides an image of the complete protein pattern on the blot. This information helps determine transfer efficiency and molecular weight of the transferred proteins. This information can also be used to determine relative quantity of sample that was loaded in each lane.
Several total protein stains are available and commonly used in western blotting. The table below provides an overview of total protein detection methods and applications.
Total Protein Detection Methods for Western Blotting
| Detection Method | Sensitivity | Advantages | Disadvantages | Imaging |
|---|---|---|---|---|
| Anionic dyes (Ponceau S, Fast Green, Coomassie Brilliant Blue, Amido black) | 100–1,000 ng | Inexpensive, rapid | Coomassie and Amido black are not compatible with downstream immunodetection | Epi illumination |
| Fluorescent stains (SYPRO Ruby and Deep Purple) | 2–8 ng | Sensitive; most are compatible with immunodetection | Additional staining and destaining steps | Fluorescent capable digital imager with UV, visible light LED, or lasers |
| Stain-Free Imaging | 2–28 ng | Rapid and convenient—ready in less than one minute, and no separate staining or destaining required. Sensitivity comparable to Coomassie | Requires use of Stain-Free Gels and compatible imager | Bio-Rad imagers |
Stain-Free Imaging Technology
Bio-Rad's stain-free technology allows direct visualization, analysis, and documentation of protein samples in PAGE gels and on blots, without staining or destaining. Stain-free imaging provides equal or better sensitivity compared to Coomassie staining and eliminates organic waste disposal concerns.
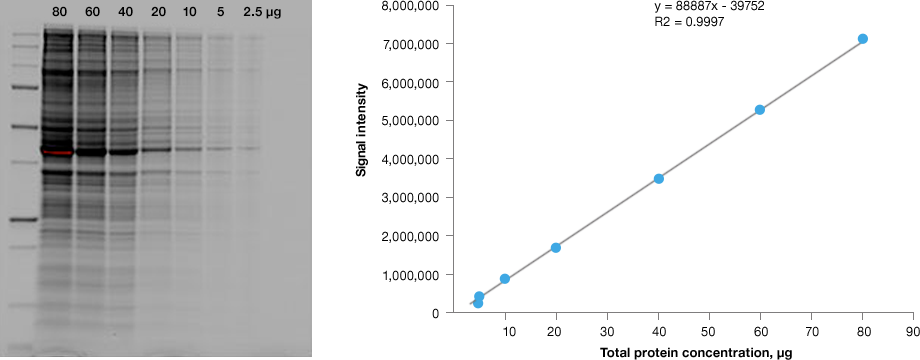
Linear dynamic range provided by stain-free technology for total protein measurements. HeLa cell lysate dilutions from 80–2.5 µg total protein.
Learn More about Stain-Free Imaging Technology »
Total Protein Normalization
Bio-Rad’s stain-free technology can be used for total protein normalization of western blots. Total protein normalization uses the signal of all proteins in a sample to determine load. This approach is more robust against expression changes of any one protein to an experimental condition, which can occur when using a single “housekeeping” protein.
Immunodetection Tips and Protocols
Western Detection Tips

Use high-quality primary antibodies.
A good antibody is sensitive, meaning it can detect low amounts of your target, and is also specific, recognizing only the target, without giving spurious secondary bands. Antibody vendors are increasingly offering antibodies that have been certified for western blotting.

Use cross-adsorbed secondary antibodies.
Cross-adsorbed antibodies offer a lower chance of cross-reactivity and reduced background giving more reliable results. They are readily commercially available so they should be used whenever possible.
Optimize antibody concentration to maximize signal to background.
Sensitive detection of your target depends on high signal and low background. Reducing antibody concentration can reduce desired signal, but the greater reduction of background more than makes up for this.

For multiplex detection, first optimize antibodies to targets individually.
When detecting multiple targets simultaneously, carry out detection of each target individually first. This will simplify any needed troubleshooting.
Western Detection Protocols and Resources

Find the right products for you using the free Western Blot Selector Tool

Western Blotting Protocol Library
Filter by your laboratory set-up and reagents to get a custom western blotting protocol that best fits your needs.

Stain-Free Western Blotting Guide
Find out how Stain-Free technology can revolutionize your western blotting.

Better Western Blotting Guide
Tips, Techniques, and Technologies from the Western Blotting Experts at Bio-Rad Laboratories.

Protein Blotting Guide
(PDF 7.2 MB)Details on blotting technology, methods, products, tips, techniques, and troubleshooting guidelines.

Western Blot Doctor
Our self-help troubleshooting guide covers solutions to many common and not-so-common western blotting issues and helps your blots look their best.

Best Practice for Western Blot Detection of Phosphorylation Events
Ten tips to ensure robust data generation and cleaner blots.

Protocol: Detection of Phosphorylated Proteins by Western Blotting
This protocol describes how to detect phosphorylated proteins by western blotting using Phospho-Specific PrecisionAb Antibodies.

Fundamentals of Western Blotting Course #3: Immunodetection

Request your Free Electrophoresis & Western Blotting Layout Post-It
This simple tool allows users to keep track of their Western Blotting experiment from sample preparation to imaging.

Get Your Free Protein Standard Temporary Tattoo
You too can sport a Precision Plus Protein Kaleidoscope standard tatto temporarily.

Western Blot University
Courses designed to make you a western blotting expert.