
Better Electrophoresis
Theory and advice for the best separation.
On This Page | Electrophoresis Basics | Gel Types | Buffer & Gel Systems | Protein Standards | Power Settings | Electrophoresis Tips | Protocols & Resources |
Sign Up for Western Blotting Tips and Webinar Alerts
Electrophoresis Basics
How Protein Electrophoresis Works
The term electrophoresis refers to the movement of charged molecules in response to an electric field. Electrophoresis has been developed into a standard molecular biology laboratory technique, where molecules of different sizes and charges are transported through a medium at different rates by an applied electrical field, resulting in their separation.
In an electric field, proteins move toward the electrode of opposite charge. Proteins have a wide range of sizes and shapes and have charges imparted to them by their constituent amino acids. As a result, proteins have characteristic migration rates that can be exploited for separation. The total charge on a protein in an aqueous medium depends on the degree of ionization of its acid and base groups, which can be controlled by buffering. Because the mass of a protein is constant, its charge-to-mass ratio is thereby altered. Protein conformation can also be changed by buffering, detergents, and oxidation/reduction. These factors allow for differences in protein migration rates to be tuned to a degree, thus enhancing separation. In protein electrophoresis, the proteins move through and aqueous polymer gel, which retards migration and retains them for imaging and analysis. The rate proteins move in gel electrophoresis is governed by relationships among the characteristics of the electrophoresis system and the proteins.
Factors affecting protein migration include:
- Gel composition and strength (weight percentage of polymer)
- Electric field strength
- Temperature
- Protein size, shape, and charge — which are in turn affected by
- Buffer pH, ion type, and concentration
- Addition of detergents or reducing agents
Polyacrylamide Gel Electrophoresis (PAGE) Overview
Almost all methods for protein separation use an aqueous polyacrylamide gel as a size-selective sieve during separation. This polyacrylamide gel is what lends the name to the technique: polyacrylamide gel electrophoresis, or PAGE. As proteins move through the gel in response to an electric field, the gel’s pore structure allows smaller proteins to travel more rapidly than larger proteins. This difference in migration rate between smaller and larger proteins leads to their physical separation in the gel.
In most PAGE applications, the gel is mounted between two buffer chambers, and the only electrical path between the two buffers is through the gel. Usually, the gel has a vertical orientation, and the gel is cast with a comb that creates wells in which the samples are deposited. Applying an electrical field across the buffer chambers forces the migration of proteins into and through the gel. The movement of the proteins through the gel is normally tracked visually by the addition of a dye to the sample, which can be seen moving further down the gel over time. If the gel is run for too long, the proteins can run off the gel.
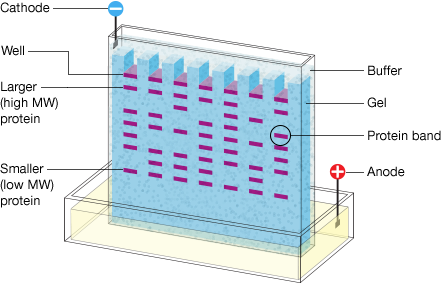
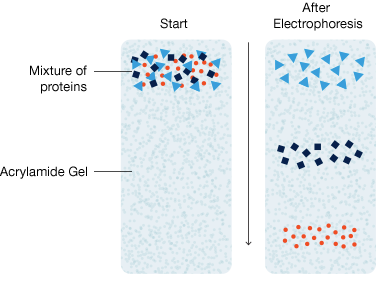
Schematic of electrophoretic protein separation in a polyacrylamide gel. MW, molecular weight.
Native PAGE
Native PAGE is a method in which proteins are prepared in nonreducing, nondenaturing sample buffer, and electrophoresis is also performed in the absence of denaturing and reducing agents. Since the native charge-to-mass ratio is preserved, protein mobility is determined by a complex combination of factors. Additionally, since protein-protein interactions are retained during separation, some proteins may also migrate together as multisubunit complexes and move in unpredictable ways. Moreover, because the native charge is preserved, proteins can migrate towards either electrode depending on their charge.
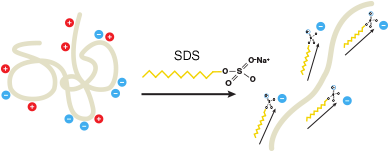
Effect of SDS on proteins in aqueous buffer. SDS helps denature proteins so they adopt a rod-like conformation. SDS also adds an overall negative charge to the protein.
Migration of Proteins and Ions

Denatured proteins are loaded into the wells.

Voltage applied and proteins move into gel. Chloride ions present in gel (leading ions) run faster than SDS-bound proteins and form an ion front. Glycinate ions (trailing ions) flow in from running buffer and form a front behind proteins.

Proteins are stacked between chloride and glycinate ion fronts. Percentage of acrylamide increases and the pore size decreases at interface between stacking and resolving gels. Movement of proteins into the resolving gel is met with increased resistance.

Proteins are stacked between chloride and glycinate ion fronts. Percentage of acrylamide increases and the pore size decreases at interface between stacking and resolving gels. Movement of proteins into the resolving gel is met with increased resistance.

The smaller pore size of the resolving gel separates the proteins. Since the charge-to-mass ratio is equal in all the proteins of the sample, separation is based on molecular weight only.
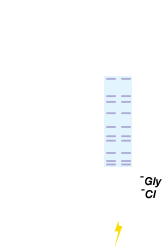
Individual proteins are separated into bands according to their molecular weights.
SDS-PAGE
This method incorporates the detergent sodium dodecyl sulfate (SDS) into the buffer system and has become the most popular form of protein electrophoresis. When proteins are separated in the presence of SDS and denaturing agents, for example, reducing agents to dissociate intra-protein disulfide bonds, they become fully denatured and dissociate from each other. In addition, SDS binds noncovalently to proteins and imparts several characteristics that can be exploited for protein separation:
- Since SDS is negatively charged, it masks the intrinsic charge of the protein and provides an overall negative charge to the proteins
- A similar charge-to-mass ratio for all proteins in a mixture, since SDS binds at a consistent rate of 1.4 gram of SDS per gram of protein
- A long, rod-Iike shape of the proteins instead of a complex tertiary conformation
As a result, the rate at which SDS-bound protein migrates in a gel depends primarily on its size, enabling molecular weight estimation of the individual proteins as they are resolved into separate bands on the gel.
Types of Protein Gels
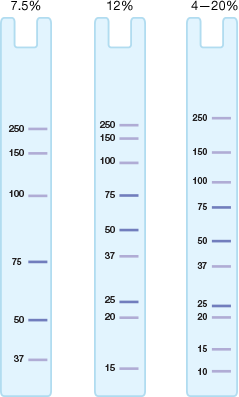
Resolution of proteins of various sizes on gels with different percentages of polyacrylamide. High-molecular-weight proteins are better resolved with low-percentage acrylamide gels, whereas low-molecular-weight proteins are best resolved with a high percentage of acrylamide. Gradient gels are best for protein samples with a wide range of molecular weights.
Gel Preparation
Polyacrylamide gels are prepared in the laboratory from a solution of acrylamide monomer, which can polymerize into long chains, and bis-acrylamide, which can form cross-links between chains. Polymerization occurs with the addition of an activator, usually the oxidizing agent ammonium persulfate (APS), which provides a source of free radicals to initiate polymerization, typically along with a catalyst to promote radical formation, commonly tetramethylethylenediamine (TEMED).
Gel Percentage Selection
Gel percentage is the main determinant of protein migration rates in PAGE. Polyacrylamide gels are characterized by two parameters: total monomer concentration (%T, in g/100 ml) and weight percentage of cross-Iinker (%C). By varying these two parameters, the pore size of the gel can be optimized to yield the best separation and resolution for the proteins of interest. %T indicates the relative pore size of the resulting polyacrylamide gel; a higher %T refers to a larger polymer-to-water ratio and smaller average pore sizes.
The practical ranges for monomer concentration are stock solutions of 30–40%, with different ratios of acrylamide monomer to cross-linker. The designations 19:1, 29:1, or 37.5:1 on acrylamide/bis solutions represent cross-Iinker ratios of 5%, 3.3%, and 2.7%, respectively, with 37.5:1 being the most commonly used ratio for protein separations.
To see how protein bands of various sizes resolve on gels with different gel percentages and buffer chemistry:
See Our Protein Gel Migration Charts »
Single-Percentage vs. Gradient Gels
Gels can be made with a single, continuous percentage throughout the gel (single-percentage gels), or they can be cast with a gradient of total monomer concentration, %T, through the gel (gradient gels). Gel percentages for single-percentage gels are usually between 7.5% and 20%, and for typical gradient gels are 4–15% and 10–20%. Use protein migration charts and tables to select the gel type that offers optimum resolution of your sample.
Gel Percentage Selection Guide
| Molecular Weights of Proteins in Your Sample | SingIe-Percentage or Gradient Gel? | Applications |
|---|---|---|
| Narrow range | Single-percentage | Use singIe-percentage gels to separate bands that are close in molecular weight. Since optimum separation occurs in the lower half of the gel, choose a percentage in which your protein of interest migrates to the lower half of the gel; higher percentage for small proteins, lower percentage for large proteins. |
| Broad range | Gradient | Use gradient gels to separate samples containing a broad range of molecular weights. Gradient gels allow resolution of both high- and low-molecular-weight bands on the same gel. The larger pore size toward the top of the gel permits resolution of larger molecules, while pore sizes that decrease toward the bottom of the gel restrict excessive separation of small molecules. |
| Unknown range | Gradient then single-percentage | For new or unknown samples, first try a broad gradient such as 4–20% or 8–16%, for a global evaluation of the sample. Then use an appropriate singIe-percentage gel once a particular size range of proteins has been identified. |
To select the best gel percentages and buffer chemistry for your samples:
Precast Gels vs. Handcast Gels
Handcast gels must be prepared from acrylamide and bis-acrylamide monomer solutions; the component solutions are prepared, mixed together, and then poured between two glass plates to polymerize. Because acrylamide and bis-acrylamide are neurotoxins, care must be taken to avoid direct contact with the solutions and to clean up any spills, and the acrylamide powders present an inhalation hazard. In addition, the solutions must be completely degassed to avoid the formation of bubbles in the gel. Overall, the hand-casting process is labor intensive, is not as controlled as it is by gel manufacturers and contributes to more irregularities and less reproducibility than precast gels.
| 
| |
|---|---|---|
| Convenience | Ready to use | Requires preparation and mixing of acrylamide solutions and pouring of gels |
| Consistency and reproducibility | Gel manufacturers tightly control process and gel quality leading to more consistent results. | Hand pouring of individual gels can lead to inconsistent gel quality that can lead to inconsistent results. |
| Shelf life | Formulations lasting up to 1 year | 4 weeks or less |
| Cost | Can be 2–3x more than hand-casting. | Low-cost |
See Our Protein Gels For Electorphoresis And Western Blot »
Stain-Free Gels

This is a new category of polyacrylamide gel which behaves like a single percentage or gradient gel but contains a unique trihalo compound. This compound reacts with the tryptophan residues in a UV-induced reaction to produce fluorescence, which can be easily detected by a compatible imager either within gels or on a membrane. Activation of the trihalo compounds in the gel adds 58 Da moieties to available tryptophan residues and is required for protein visualization. Because the compound becomes covalently bound to the protein molecules, they can be imaged repeatedly on the gel or on a membrane after transfer, without additional staining and destaining steps. The sensitivity of the system is comparable to staining with Coomassie (Brilliant) Blue for proteins with a tryptophan content >1.5%; sensitivity superior to Coomassie staining is possible for proteins with a tryptophan content >3%.
Learn More about Stain-Free Gel Technology »

Free eBook
Find out how Stain-Free technology can revolutionize your western blotting
Stain-Free Western Blotting
Stain-Free gel technology allow users to instantaneously visualize protein after electrophoresis, verify that all proteins have been transferred out of the gel, and validate that all of the proteins are on the membrane without the need for additional staining/destaining steps. This potentially saves time wasted on western blots with problems that would not otherwise have been detected until the later stages of blot processing and development.
The Stain-Free Western Blotting Workflow — Faster Results, Better Data
1
Visualize gel immediately after electrophoresis (5 minutes)
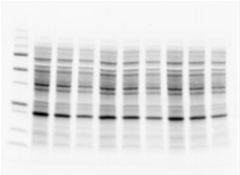
Pre-transfer gel stain-free image to check sample integrity and separation quality.
2
Verify that all proteins have been transferred out of the gel

Post-transfer gel stain-free image to measure transfer efficiency.
3
Assess transfer efficiency onto the membrane before detection
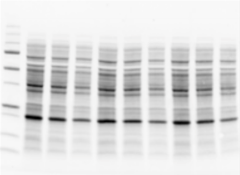
Stain-free blot image as loading control.
Learn More about Our Proprietary Stain-Free Western Blotting Workflow »
Electrophoresis Buffer Systems and Gel Chemistries
The pH and ionic composition of the buffer system determine the power requirements and heavily influence the separation characteristics of a polyacrylamide gel. Buffer systems include the buffers used to:
- Cast the gel
- Prepare the sample (sample buffer)
- Fill the electrode reservoirs (running buffer)
Most common PAGE applications utilize discontinuous buffer systems, where two ions differing in electrophoretic mobility form a moving boundary when a voltage is applied. Proteins have an intermediate mobility making them stack, or concentrate, into a narrow zone at the beginning of electrophoresis. As that zone moves through the gel, the sieving effect of the gel matrix causes proteins of different molecular weights to move at different rates. Varying the types of ions used in the buffers changes the separation characteristics and stability of the gel.
Gel and Buffer Chemistries for PAGE
Buffers | Precast (Format) Gels | |||||
|---|---|---|---|---|---|---|
| Gel Type | Selection Criteria | Sample | Running | Mini-PROTEAN | Criterion | Handcast |
| SDS-PAGE | ||||||
| Tris-HCI, pH 8.6 | Easy to prepare, reagents inexpensive and readily available; best choice when switching between precast and handcast gels and need to compare results | Laemmli | Tris/glycine/SDS | • | • | • |
| TGX | Laemmli-like extended shelf-life gels; best choice when long shelf life is needed (Mini-PROTEAN) and traditional Laemmli separation patterns are desired | Laemmli | Tris/glycine/SDS | • | • | Bio-Rad FastCast |
| TGX Stain-Free | Laemmli-like extended shelf-life gels with trihalo compounds for rapid fluorescence (Mini-PROTEAN) detection without staining | Laemmli | Tris/glycine/SDS | • | • | — |
| Bis-Tris, pH 6.4 | Offer longest shelf life, but reagents may be costly | XT | XT MOPS or XT MES | — | • | — |
| Tris-acetate, pH 7.0 | Best resolution of high-molecular-weight proteins; use for peptide sequencing or mass spectrometry applications | XT Tricine or Tris/Tricine/SDS | — | • | • | — |
| Native PAGE | ||||||
| TGX | Laemmli-like extended shelf-life gels; best choice when long shelf life is needed and traditional Laemmli separation patterns are desired | Native | Tris/glycine | • | • | — |
| Stain-Free | Laemmli-like gels with trihalo compounds for rapid fluorescence detection without staining | Native | Tris/glycine | — | • | — |
| TGX Stain-Free | Laemmli-like extended shelf-life gels with trihalo compounds for rapid fluorescence detection without staining | Native | Tris/glycine | • | • | — |
| Tris-acetate, pH 7.0 | Offer best separation of high-molecular-weight proteins and protein complexes | Native | Tris/glycine | — | • | • |
| Peptide Analysis | ||||||
| Tris-Tricine | Optimized for separating peptide and proteins with molecular weight <1,000 | Tricine | Tris/Tricine/SDS | • | • | • |
| Isoelectric Focusing (IEF) | ||||||
| IEF | Cast with Bio-Lyte Ampholytes, allow separation by protein pI; contain no denaturing agents, so IEF is performed under native conditions | 50% glycerol | IEF cathode and IEF anode buffers | • | • | — |
Protein Electrophoresis Standards
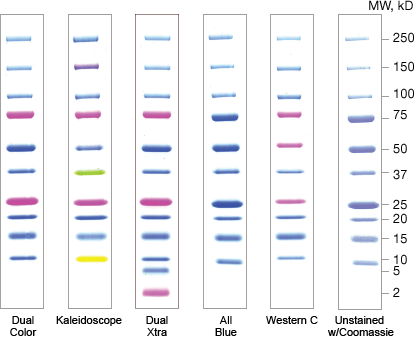
Protein standards are mixtures of well-characterized or recombinant proteins that are loaded alongside protein samples in a gel. They are used to monitor separation as well as estimate the size and concentration of the proteins separated in a gel.
Select protein standards that offer:
- Good resolution of the proteins in the size range of interest
- Compatibility with downstream analysis (for example, blotting)
Protein standards are available as prestained or unstained sets of purified natural proteins or recombinant proteins. Prestained standards allow easy and direct visualization of their separation during electrophoresis and their subsequent transfer to membranes. Although prestained standards can be used for size estimation, unstained protein standards will provide the most accurate size determinations.

Recombinant Standards
- Engineered proteins, tight bands
- Evenly spaced
- Precise MW estimations, confirmed by mass spectrometry
- Affinity tags for detection
Recombinant standards are engineered to display specific attributes such as evenly spaced molecular weights or affinity tags for easy detection. These standards often offer absolute molecular weight accuracy confirmed by mass spectrometry. Because they contain a known amount of protein in each band, they also allow approximation of protein concentration.
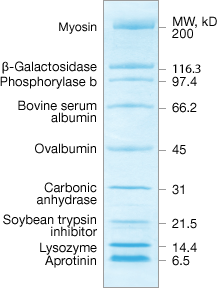
Natural Standards
- Proteins bind to the dye in varying amounts at different sites producing broader bands
- Spacing based on MW of natural protein
Natural standards are blended from naturally occurring proteins. Although prestained natural standards are effective for monitoring gel separation and efficiency, they covalently bind the dye in different amounts and at different locations. This may produce broader bands than are seen with recombinant standards, making them less desirable for molecular weight estimations.
Electrophoresis Power Settings
Power supplies that are used for electrophoresis hold one parameter constant (either voltage, current, or power). The resistance, however, does not remain constant during a run: resistance changes as discontinuous buffer ion fronts move through the gel. In SDS-PAGE, resistance increases as the run progresses.
Depending on the buffer and which electrical parameter is held constant, heating of the gel may increase or decrease over the period of the run. Use external cooling during long, unsupervised runs. Temperature-controlled runs yield more uniform and reproducible results.
Constant Voltage
If the voltage is held constant throughout a separation, the current and power (heat) decrease as the resistance increases. This leads to increased run times, which allow the proteins more time to diffuse. But this is offset by the temperature-dependent decrease in diffusion rate as temperature decreases. When running multiple gels in parallel, use of constant voltage is often preferred because a single voltage can be specified that is independent of the number of gels run in parallel.
Constant Current
If the current is held constant during a run, the voltage, power, and consequently the heat of the gel chamber increases during the run. Constant current conditions result in shorter but hotter runs than constant voltage runs. This increase in heat increases the rate of diffusion of the proteins, leading to fuzzier bands unless additional cooling is supplied.
Constant Power
Holding the power constant minimizes the risk of overheating. Many power supplies do not offer this mode of power control and gel manufacturers do not often provide recommended settings for constant power.
Electrophoresis Tips

Wash out wells.
Prior to loading sample in the gel, wash out each well with a pipet loaded with running buffer from the upper buffer chamber. This helps wash out any bits of polymerized acrylamide as well as residual acrylamide casting solution. This helps avoid misshapen band morphology.%20not%20%20straight.jpg)
Avoid excess gel heating.
If a gel runs too hot during electrophoresis, bands may “smile,” where the band pattern curves upward at both sides of the gel. Excessive heating can occur if buffer composition is incorrect, not mixed well, or if power conditions are excessive.
Ensure sample buffer ionic strength is correct.
Both excess salts in samples and (conversely), an ionic strength of the sample that is lower than the gel, can lead to bands appearing skewed or distorted.Do not overload samples.
It is easier to detect rare protein targets if more sample is loaded on a gel. However, excessive loads can lead to vertical streaking during electrophoresis. With heavy sample loads, reducing electrophoresis voltage by 25% may help to minimize streaking.
Quick Tips:
How to Minimize Band Distortion During Gel Electrophoresis for Western Blotting
Watch the video below to learn how to eliminate smiling and frowning during electrophoresis to get the best gel resolution for your western blots.
Electrophoresis Protocols and Resources

Find the right products for you using the free Western Blot Selector Tool

Western Blotting Protocol Library
Filter by your laboratory set-up and reagents to get a custom western blotting protocol that best fits your needs.

Better Western Blotting Guide
Tips, Techniques, and Technologies from the Western Blotting Experts at Bio-Rad Laboratories

Protein Electrophoresis Guide
(PDF 8.35 MB)A guide containing electrophoresis theories and techniques plus tips on troubleshooting common problems using Bio-Rad products.

Little Book of Standards
(PDF 5.12 MB)Complete reference information for all of Bio-Rad’s protein standards and nucleic acid standards.

Western Blot Doctor Troubleshooting Guide
Our self-help troubleshooting guide covers solutions to many common and not-so-common western blotting issues and helps your blots look their best.

Fundamentals of Western Blotting Course #2: Gel Electrophoresis and Transfer

Request your Free Electrophoresis & Western Blotting Layout Post-It
This simple tool allows users to keep track of their Western Blotting experiment from sample preparation to imaging.

Get Your Free Protein Standard Temporary Tattoo
You too can sport a Precision Plus Protein Kaleidoscope standard tatto temporarily.

Western Blot University
Courses designed to make you a western blotting expert.