
Proper experimental design is the key to any gene expression study. Since mRNA transcription can be sensitive to external stimuli that are unrelated to the processes studied, it is important to work under tightly controlled and well-defined conditions. Taking the time to define experimental procedures, control groups, type and number of replicates, experimental conditions, and sample handling methods within each group is essential to minimize variability. Each of these parameters should be carefully recorded prior to conducting real-time PCR experiments to ensure good biological reproducibility for published data.
Related Topics: Gene Expression Analysis.
Page Contents
For quantifying gene expression, sample material should be as homogeneous as possible. If your tissue sample consists of many different cell types, pinpointing the expression pattern of your target gene may be difficult. If you have a heterogeneous sample, use one of the many methods available for separating and isolating specific cell types. These methods include tissue dissection, needle biopsies, and laser capture microdissection. The collected cells can then be used to obtain the RNA samples.
Either total or poly(A+) RNA can be used for most real-time RT-qPCR applications. One critical consideration in working with RNA is to avoid RNases in your solutions, consumables, and labware. Ready-to-use solutions that are RNase free can be purchased. Alternatively, treat solutions with diethyl pyrocarbonate (DEPC), and then autoclave them. RNases on labware can also be inactivated by DEPC treatment, or by baking at 250°C for 3 hr.
Prepared RNA samples may need DNase treatment to prevent potential amplification of any contaminating genomic DNA, which could lead to overestimation of the copy number of an mRNA. When starting material is limited, however, DNase treatment may be inadvisable, because the additional manipulation could result in loss of RNA. The amplification of potential contaminating genomic DNA can be precluded by designing transcript-specific primers, for example, primers that span or amplify across splice junctions.
Accurate nucleic acid quantification is essential for gene expression analysis, especially if total RNA amounts are used to normalize target gene expression. RNA concentrations and purity are commonly determined by measuring the ratio of the UV absorbance at 260 nm and 280 nm. The overall sensitivity of this method is low, especially for relatively dilute samples, and it doesn't indicate the quality of the RNA. To determine the quality and quantity of your sample, we recommend using the Experion™ automated electrophoresis system. Examples of a high-quality total RNA preparation and a poor-quality total RNA preparation are shown in Figure 1.
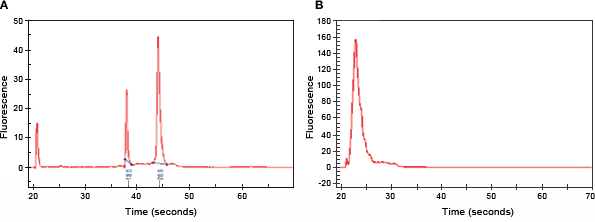
Fig. 1. RNA quality assessed by an Experion automated electrophoresis system. A, an example of a high-quality total RNA preparation showing distinct peaks for the 18S and 28S rRNAs; B, an example of poor-quality total RNA preparation where only small, degraded fragments are observed.
In qPCR experiments, reference genes are used as controls to normalize the data by correcting for differences in quantities of cDNA used as a template. A perfect reference gene is therefore one that does not exhibit changes in expression between samples from various experimental conditions or time points. Several genes, such as GADPH, ACTB, or 16S rRNA, are often used as reference genes. However, a number of studies have indicated that the expression of these genes may vary considerably between tissues or between treatments, and this variability may make them unsuitable for use as reference genes. Reference genes must therefore be carefully selected based on experimental data. A good reference gene should have an M value below 0.5 in homogeneous sample sets, and below 1 in heterogeneous sample sets (Vandesompele et al. 2002). Typically, between three and five good reference genes are required to achieve the most accurate normalization.
Relative quantification is used to compare the amount of a target nucleic acid in equivalent amounts of different samples.
Absolute quantification is used to determine how much (number of copies, ng, etc.) of a target gene is present in a particular sample without reference to other samples. Absolute quantification is conceptually simple and the mathematical calculations are easy to perform. It involves comparing the quantification cycle (Cq) values of test samples to those of standards of known quantity plotted on a standard curve. Usually, the quantity is normalized to a unit amount of sample, such as number of cells, volume, or total amount of nucleic acid. To use the method, you must have a reliable source of template of known concentration, and standards must be amplified in parallel with the samples every time the experiment is performed.
After the Cq values are measured, different methods can be used to determine the expression level of the target gene in the test sample relative to the calibrator sample.
The 2-ΔΔCq (Livak) Method
The Livak method for relative gene expression analysis is widely used and easy to perform. This method assumes that both target and reference genes are amplified with efficiencies near 100% and within 5% of each other. Before using the Livak method, it is essential to verify these assumptions by determining the amplification efficiencies of target and the reference genes.
Once you have established that the target and reference genes have similar and nearly 100% amplification efficiencies, you can determine the relative difference in expression level of your target gene in different samples using the steps below:
- Normalize the Cq of the target gene to that of the reference (ref) gene for both the test sample and the calibrator sample:
ΔCq(test) = Cq(target, test) - Cq(ref, test)
ΔCq(calibrator) = Cq(target, calibrator) - Cq(ref, calibrator) - Normalize the ΔCq of the test sample to the ΔCq of the calibrator:
ΔΔCq = ΔCq(test) - ΔCq(calibrator) - Calculate the expression ratio:
2-ΔΔCq = normalized expression ratio
The result is the ratio of the target gene in the test sample to the calibrator sample, normalized to the expression of the reference gene. Normalizing the expression of the target gene to that of the reference gene compensates for any difference in the amount of sample tissue.
If the target and the reference genes do not have similar amplification efficiencies, you can either optimize or redesign the assays, or you can use the Pfaffl method. If, on the other hand, the target and the reference genes have an identical amplification efficiency, but the efficiency is not equal to 2, a modified form of the Livak method may be used by replacing the 2 in the equation by the actual amplification efficiency. For example, if the amplification efficiency of both the target and the reference gene is 1.95, the following equation should be used:
Ratio = 1.95-ΔΔCq
The ΔCq Method Using a Reference Gene
The ΔCq method using a reference gene is a variation of the Livak method that is simpler to perform and gives essentially the same results. In contrast to the ΔCq values obtained with relative quantification normalized against a unit mass, this method uses the difference between reference and target Cq values for each sample. Despite the simplicity of the approach, it is indeed normalized expression. The expression level of the reference gene is taken into account. The key difference in the results is that the expression value of the calibrator sample is not 1.0. If the resulting expression values obtained in this method are divided by the expression value of a chosen calibrator, the results of this calculation are exactly the same as those obtained with the Livak method:
Ratio (reference/target) = 2Cq(ref) - Cq(target)
The mathematical assumptions for this approach are the same as those for the Livak method.
The Pfaffl Method
The Livak method for calculating relative gene expression is valid only when the amplification efficiencies of the target and reference genes are similar. If the amplification efficiencies of the two PCR products are not the same, an alternative formula must be used to determine the relative expression of the target gene in different samples. To determine the expression ratio between the test sample and calibrator for a target normalized to a reference (ref), use the following equation:
Ratio = (Etarget)ΔCq, target (calibrator - test)/(Eref)ΔCq, ref (calibrator - test)
where
| Etarget | is the amplification efficiency of the target gene. |
| Eref | is the amplification efficiency of the reference gene. |
| ΔCq, ref (calibrator - test) | is the Cq of the reference gene in the calibrator minus the Cq of the reference gene in the test sample. |
| ΔCq, target (calibrator - test) | is the Cq of the target gene in the calibrator minus the Cq of the target gene in the test sample. |
The above equation assumes that each gene (target and reference) has the same amplification efficiency in test samples and calibrator samples, but it is not necessary that the target and reference genes have the same amplification efficiency.
In order to understand the significance of the data that have been collected and analyzed, statistical values need to be determined. The specific measure that is used will depend on how the data are distributed (for example, "normal," symmetrical), the sample size, the number of factors being considered, etc. A tool that can assist with statistical analysis is qbase+ software (Biogazelle, Belgium), which incorporates a guided statistical wizard that aids setup.
Real-time quantitative PCR (qPCR) has become a definitive technique for quantitating differences in gene expression levels between samples. Over the past 10 years, the popularity of this method has grown exponentially, with the publication of well over 25,000 papers making reference to qPCR data, from diverse fields of science including agricultural, environmental, industrial, and medical research. Apart from the broad applicability of the technique, one of the central factors that has stimulated this impressive growth is the increased demand from journal review panels for the use of qPCR to support phenotypic observations with quantitative, molecular data. Furthermore, gene expression analysis is now being used to support protein expression data from proteomics-based assays. The biotechnology industry has responded to the rapid adoption of this technique by developing reagents and instruments to perform qPCR experiments. However, without strict guidelines to follow, researchers generally designed their experiments based on information gathered from disparate sources, and this resulted in data of variable quality. A number of technical deficiencies can affect qPCR assay performance, including improper experimental design, inadequate controls and replicates, lack of well-defined experimental conditions and sample handling techniques, poor quality of RNA samples, suboptimal choice of primers for reverse transcription (RT) and qPCR reactions, lack of validation of reference genes, and inappropriate methods of data analysis.
To assist the scientific community in producing consistent, high-quality data from qPCR experiments, guidelines for the minimum information for publication of quantitative real-time PCR experiments (MIQE) have been recently published (Bustin et al. 2009).This has been followed by the development of an XML-based real-time PCR data markup language (RDML) by the RDML consortium (Lefever et al. 2009) for the consistent reporting of real-time PCR data. This consortium is active in the development of appropriate and standardized terminology, guidelines on minimum information for biological and biomedical investigations, and a flexible and universal data file structure with tools to create, process, and validate RDML files (www.rdml.org).
The ultimate goal of RDML and MIQE guidelines is to establish a clear framework within which to conduct RT-qPCR experiments and to provide an established yardstick for reviewers and editors to use in the evaluation of the technical quality of submitted manuscripts. As a consequence, investigations that use this widely applied methodology will produce data that are more consistent, more comparable, and ultimately more reliable.
Bustin SA et al. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–622. PMID: 19246619
Lefever S et al. (2009). RDML: Structured language and reporting guidelines for real-time quantitative PCR data. Nucleic Acids Res 37, 2065–2069. PMID: 19223324
Vandesompele J et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, research0034.1. PMID: 12184808
Guénin S et al. (2009). Normalization of qRT-PCR data: The necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot 60, 487–493. PMID: 19264760
Kitchen RR et al. (2010). Statistical aspects of quantitative real-time PCR experiment design. Methods 50, 231–236. PMID: 20109551
Yuan JS et al. (2006). Statistical analysis of real-time PCR data. BMC Bioinformatics 7, 85. PMID: 16504059