

The ProteOn XPR36 system detects binding between a ligand immobilized on a sensor chip and an analyte flowing over it in a microfluidic channel using surface plasmon resonance (SPR) technology. The real-time data generated by the ProteOn XPR36 System is analyzed using the ProteOn Manager Software which comes with several different binding models for analyzing kinetic data, ranging from a simple Langmuir model of 1:1 binding to more complex binding models such as Langmuir binding with mass transport limitations. The ProteOn system is especially adept at obtaining kinetic data as the novel XPR technology allows for the creation of 6 x 6 interaction array on the ProteOn sensor chips. Based on it, the patented One-shot Kinetics approach is realized by the interrogation of six ligands with a series of up to six concentrations of one analyte in a single injection. In addition to investigating the kinetics of an interaction, the ProteOn can be used to characterize epitopes on an antigen, investigate binding thermodynamics, screen small molecules for affinity, or to determine the active concentration of an analyte in a heterogeneous sample.
Related Topics: ProteOn Sensor Chips.
Page Contents
The ProteOn system uses SPR to detect the interaction between two unlabeled biomolecules in real-time. A typical ProteOn experiment involves immobilizing a ligand (or target) to a functionalized gold sensor chip and flowing an analyte (another protein or small molecule) over the chip surface to investigate the binding affinity and kinetics of the analyte to the ligand. Binding of analyte to the ligand is tracked by following the change of SPR signal measured at the surface of the sensor chip over time (Figure 1).
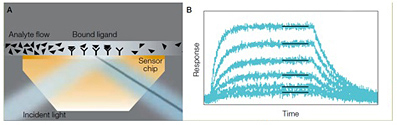
Fig. 1. Schematic illustration of the conversion of shift in SPR angle to sensorgrams. A, as analyte binds to ligand molecules on the sensor chip surface, the intensity minimum (shadow) produced by the SPR effect shifts. This intensity shift is measured in real time for 36 interaction spots and 42 interspot references. B, each of the resulting sensorgrams is fitted to an appropriate mathematical model to quantitatively characterize the interaction (shown for one set of 6).
SPR is an optical phenomenon that occurs when p-polarized light at a certain wavelength and angle is reflected off a thin metal film (the gold film coated on the sensorchip) under the condition of total internal reflection (TIR). The light excites surface plasmons in the metal at a certain incident angle. The TIR field generates an evanescent wave in the thin metal film that extends hundreds of nanometers from the surface into the medium above, in this case the molecules in contact with the chip surface. The excited surface plasmons are very sensitive to the refractive index change at the surface of the thin metal film. Thus the incident angle of the light required for SPR is impacted by the refractive index change of the molecules in contact with the chip surface. In an SPR binding experiment, this refractive index change is brought about by binding of analyte in solution to ligand immobilized on the chip surface; therefore, tracking the change in the incident angle required for SPR allows one to monitor biomolecular interactions in real-time. The change of the incident angle required for SPR is defined as SPR signal in the unit of response unit (RU). 1 RU is 1/1,000,000 of a refractive index unit, and is roughly equivalent to a surface density of protein at approximately 1 pg/mm2. For a more in-depth discussion of SPR, see a recent review that offers an overview of SPR theory and different SPR configurations (Daghestani et al. 2010).
Plotting the SPR signal over time during the interaction between an analyte and a ligand results in a sensorgram, a visual representation of the interaction over time. Figure 2 shows an example sensorgram for an antibody-antigen interaction. The binding response initially increases as analyte is flowed over the sensor chip and associates with the immobilized ligand and then decreases as the analyte solution is replaced with buffer and the binding complex dissociates. If binding equilibrium is reached during the association phase, the sensorgram will reach a constant plateau before the analyte solution is replaced with buffer and the binding complex dissociates. Fitting the sensorgram data to a binding model allows for the calculation of the association (ka) and dissociation (kd) rate constants and determination of the binding affinity. Traditionally, kinetic measurements with SPR usually involve sequential injections of analyte at increasing concentrations over the same ligand surface, which requires complete removal of the analyte, or regeneration of the ligand surface, between analyte injections. In an ideal case, regeneration of the ligand surface is seen in the sensorgram as a sharp drop in RU after dissociation and a return to the original baseline. Regeneration is usually done with a combination of dilute surfactants, salts, and acids or bases; however, care must be taken during regeneration to avoid denaturing the immobilized ligand or removing ligand from the sensor chip.
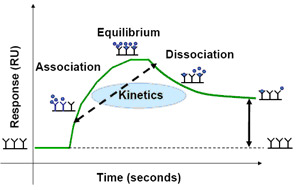
Fig. 2. Example SPR sensorgram of an antibody/antigen interaction showing the establishment of an initial baseline, an increase in response during association, a constant response after equilibrium is reached, and a decrease in response during dissociation.
The ProteOn system offers a distinct advantage over other SPR biosensor platforms because the unique 6 x 6 interaction array of the ProteOn sensor chips enable the One-shot Kinetics approach measuring the interaction of one ligand with a six-concentration series of one analyte in a single injection. This approach eliminates the need for traditional regeneration of the sensor chip between analyte injections that often deteriorates the ligand surface. Using enhanced microfluidic delivery and XPR technology, the ProteOn system can immobilize up to six separate ligands on a single sensor chip in six separate flow cells and then rotate the sensor chip 90 degrees to flow up to six separate analytes over the ligand surfaces (Figure 3).
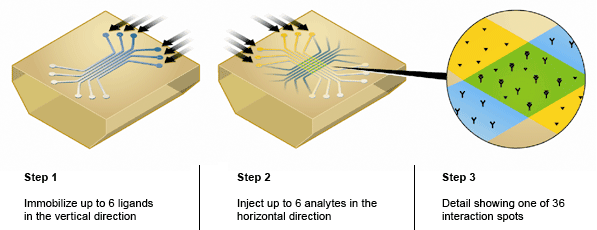
Fig. 3. ProteOn sensor chip 6 x 6 interaction array.
This unique feature of the ProteOn allows for the detection of up to 36 separate interactions on a single sensor chip and significantly increases the throughput of SPR biosensing. In a recent study, the ProteOn was used to immobilize 36 different ligands in a stepwise immobilization procedure designed for the high-throughput epitope mapping and binning of antibody/antigen interactions (Abdiche et al. 2011). The 6 x 6 interaction array of the ProteOn sensor chips also allows for in-line referencing, whereby data from unmodified spots in between the immobilized ligand spots on the sensor chip are used to subtract out artifacts such as noise and baseline drift. This in-line referencing is superior to referencing with a separate flow cell and means the ProteOn can collect high-quality SPR data at low signal-to-noise ratios, as often the case with small molecule analytes.
Using SPR, the ProteOn XPR36 system can provide a wide variety of important information on biomolecular interactions such as the specificity, affinity, qualitative ranking, kinetics, and thermodynamics of binding.
The ProteOn system can be used in pharmaceutical drug discovery, antibody characterization, immunogenicity testing, the development and manufacture of biologics, or for clinical research. However, this is not a comprehensive list and nearly any research field can benefit from using the ProteOn system if there is a need for label-free characterization of a biomolecular interaction. Key applications include:
- Quantification of binding affinity and kinetics
- Determination of binding specificity and the number of binding sites
- Characterization of the mechanism of action
- Confirmation of biomolecule binding to a target
- Screening of fragment libraries
- Validation of IC50/EC50 values during hit-to-lead optimization
- Characterization of immune responses
Some classic applications of the ProteOn are in antibody engineering. Epitopes on an antigen can be characterized by epitope mapping, a process by which the affinities of an antibody to site-directed mutants of a single antigen help pinpoint the location of an epitope. An investigation of the epitope specificity, or epitope binning, of different antibodies can be done on the ProteOn system using the sandwich assay. In this assay, a second antibody is injected over a previously-formed antigen-antibody complex to see whether or not the second antibody can still bind. Binding of the second antibody to the antigen-antibody complex, or the formation of a 'sandwich' is an indication that the second antibody recognizes a different epitope than the first antibody.
SPR can also be used to determine the active concentration of an analyte in a crude or impure sample by probing the sample of interest under mass transport control using a low flow rate and/or a high-capacity sensor chip bearing an analyte-specific ligand. Under such mass transport limited conditions the association rate of binding, or the initial binding rate, is proportional to the concentration of analyte in solution. The concentration of analyte in a crude sample can be calculated by comparing the initial binding rate to a standard curve of initial binding rates for known concentrations.
By fitting sensorgram data from a ProteOn system experiment to a suitable binding model, kinetic parameters such as the association (ka) and dissociation (kd) rate constants and the affinity (KD) can be extracted. Kinetic data are crucial for characterizing an interaction as they allow for a thorough understanding of the nuances of binding. Interactions with the same affinity (KD) can have markedly different association and dissociation rates, as seen in Figure 5. An antibody or small molecule that has a high affinity (low KD value) for a protein target may be a poor drug in-vivo if it has a very high dissociation rate and thus can be easily displaced by another molecule. This kind of information is easily obtained from an SPR experiment but would not be uncovered using a method such as isothermal calorimetry (ITC) that measures binding affinity based on binding at equilibrium. In addition, knowing the kinetics of a small molecule interaction allows for the more accurate analysis of quantitative-structure activity relationships as different structures can be evaluated by their separate effects on association and dissociation as opposed to affinity alone.
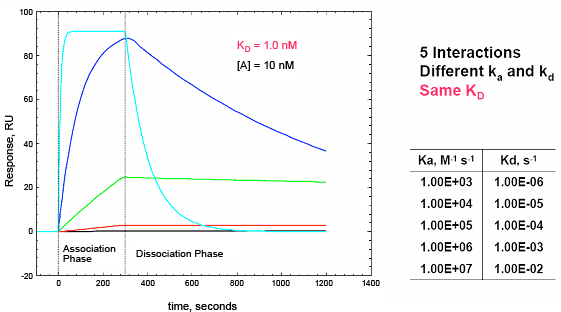
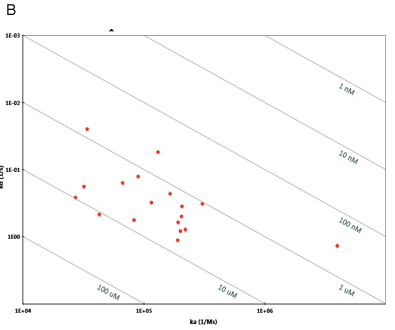
Fig. 5. A. Sensorgram plots showing the response (in RU) versus time for five different interactions with the same affinity (KD = 1.0 nM) but markedly different association (ka) and dissociation (kd) rate constants. The software model for this figure was kindly provided by Mohammed Yousef. Isoaffinity plot from ProteOn Manager™ software illustrating the importance of understanding ka, kd, in addition to KD. B. Isoaffinity plot from ProteOn Manager™ software illustrating the importance of understanding ka and kd in addition to KD. Here the affinity values cluster between 1 and 10 uM but with markedly different on and off rates.
The ProteOn Manager software gives you the option of using seven different binding models to analyze your sensorgram data:
- Langmuir: Simple 1:1 bimolecular interaction
- Simultaneous fitting of ka and kd
- Fitting of dissociation (kd) only
- Langmuir with drift: Simple 1:1 biomolecular interaction with a constant baseline drift taken into account
- Langmuir with mass transport limitations: Simple 1:1 biomolecular interaction that takes into account the rate of diffusion of analyte from the bulk to the surface
- Bivalent analyte: The analyte has two binding sites to one ligand.
- Heterogeneous analyte: Two analytes compete for binding to the same ligand site
- Heterogeneous ligand: One analyte binds to two separate ligand binding sites
- Two-State Conformation: Accounts for a change in conformation of the binding complex that occurs after the analyte binds in addition, it is possible to calculate the affinity value (KD) using equilibrium analysis, in which the equilibrium responses at different analyte concentrations are filledto a simple saturation binding model
Langmuir Model
The most commonly used binding model is the Langmuir model, which describes a simple 1:1 interaction where one ligand molecule interacts with one analyte molecule (O'Shannessy DJ et al. 1993). The complex that forms following pseudo first order kinetics and it is assumed all binding sites are equivalent and act independently of one another. In this simple 1:1 binding model, mass transport of analyte from the bulk to the surface is not taken into account. Fortunately, many interactions investigated using SPR adhere to this model and can be described the following equation:
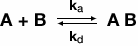
where B represents the ligand and A is the analyte. The rate of formation of the complex is governed by the association rate constant (ka) and the rate of complex dissociation is governed by the dissociation rate constant (kd). Using the above equation, one can describe the expected binding response for the association phase, equilibrium (if it is reached during the experiment), and the dissociation phase using a set of three equations. Figure 6 shows an example sensorgram for a binding interaction that can be described with a simple Langmuir model, with the relevant equations for each phase outlined.
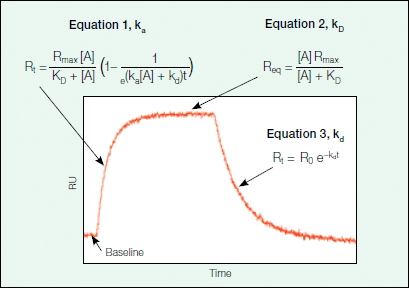
Fig. 6. The sensorgram phases.
The association phase describes the formation of the binding complex over time as analyte is flowed over the ligand surface. As analyte binds to the ligand immobilized on the sensor chip, there is an associated increase in response units as detected by the change in the SPR signal. Figure 7 provides an explanation of how Equation 1 describing the association phase (as seen in Figure 6) was derived.
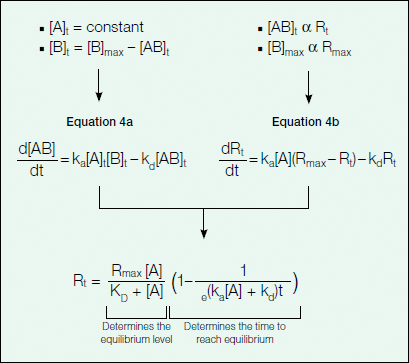
Fig. 7. Rate of complex formation.
As can be inferred from Equation 4a in Figure 7, the change in the amount of bound complex formed over time is proportional to the association (ka) and dissociation (kd) rate constants with the amount of analyte (A) in excess. Because the binding response in the sensorgram is a measure of the amount of bound complex on the sensor chip, the binding response (in RU) is proportional to the amount of bound complex and complex formation can be further described in terms of response units by Equation 4b in Figure 7. Upon integration, Equation 4b becomes Equation 1 in Figure 6, which describes the binding response with time.
During the dissociation phase, analyte is replaced with a buffer solution and the bound complex dissociates with time. This dissociation can be described by simple first order kinetics, as seen in Figure 8. When integrated, this expression becomes a simple exponential decay that describes the decrease in the binding response over time.

Fig. 8. Pre-steady-state dissociation
By fitting the relevant equations for the association and dissociation phases to the sensorgram data, one can obtain the kinetic parameters ka and kd for the association and dissociation rate constants, respectively, and the affinity (KD = kd/ka) of the interaction can be calculated. Alternatively, one can find the affinity of a given interaction by equilibrium analysis. The equilibrium binding responses at different concentrations can be fit to a simple saturation binding model, as seen in Figure 9, to extract the affinity (KD). Using this analysis method, there is no need to find kinetic parameters by fitting the association and dissociation phases to a binding model; however, the SPR data must reach equilibrium, as seen as a plateau in the sensorgram, during the experiment.
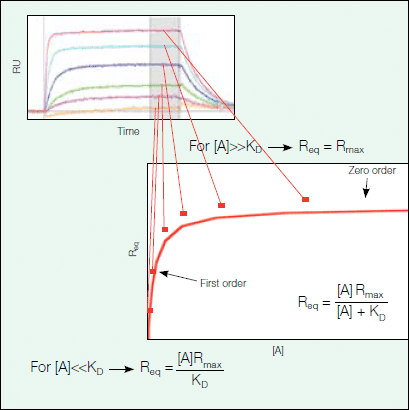
Figure 9. Steady state at equilibrium
Langmuir model with drift
The Langmuir model with drift is used when a biomolecular interaction follows simple 1:1 binding but exhibits a persistent baseline drift that interferes with data interpretation. This is applied in SPR experiments using capturing agents, as the captured ligand may leach from the surface over time. The Langmuir model with drift uses the same kinetic equations as the simple Langmiur model but calculates the drift as a linear drift with time, D*t, where D is the slope of the drift. It should be noted that this model should be applied to the experiments with slow baseline drift because fast baseline drift caused by the rapid decay of the captured ligand usually shows an exponential curvature and does not fit with this model. The optimal solution is correcting the baseline by the subtraction of a blank buffer reference (reference of blank analyte buffer over ligand surface).
Langmuir model with mass transport limitations
The Langmuir model with mass transport limitations assumes a 1:1 binding model, as is the case with the simple Langmuir model, but it takes into account the rate at which analyte is brought from the bulk solution to the sensor chip surface, which is governed by mass transfer. Some biomolecular interactions may be mass transport limited if the rate of association is faster than the rate at which analyte diffuses to the sensor chip surface. The following equation describes Langmuir binding with mass transport limitations:

where km is the mass transfer rate constant for the diffusion of analyte A from the bulk solution to the surface. A good test of whether an interaction is mass transport limited is to run the experiment at different flow rates and calculate the association rate constant. Diffusion to the surface of the sensor chip will be faster at higher flow rates; thus, if the association rate of a given interaction increases with higher flow rates and decreases with low flow rates most likely the interaction is mass transport limited. Usually one can get around a mass transport limited interaction by running the ProteOn system at high flow rates or by using low ligand density; however, there are certain situations when even these adjustments cannot eliminate the mass transport effect, and modeling the interaction using a Langmuir model with mass transport limitations is more attractive.
Bivalent analyte model
The bivalent analyte model is used when an analyte has two separate binding sites. The following equation describes binding of a bivalent analyte:
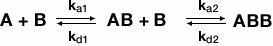
where A is the analyte and B is the ligand. The association and dissociation of the first binding event is described by ka1 and kd1, respectively, while ka2 and kd2, respectively, describe the association and dissociation of the second binding event. The first event will yield a traditional 1:1 kinetic fit where the second binding event will cause the ligand-analyte complex to stabilize, thus changing the kinetics of the reaction. Therefore, a sensorgram of a bivalent analyte binding to ligand is the result of two separate kinetic processes occurring in tandem.
Heterogeneous analyte model
When an analyte is heterogeneous, analyte may bind to the ligand in two different locations. This can occur naturally if a sample is not completely pure or if there are two different types of analyte in solution. Thus, a sensorgram of a heterogeneous analyte binding to immobilized ligand represents the sum of two separate binding interactions. If one analyte has a naturally higher affinity than the other analyte, the two may compete for binding of the ligand and the sensorgram data will reflect the binding kinetics of the higher affinity ligand. The following equations are used to describe and model the binding of a heterogeneous analyte:
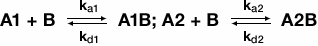
Heterogeneous ligand model
A heterogeneous ligand model assumes that there are two sites on the ligand that bind analyte. This can occur if ligand binds to the sensor chip in different orientations, resulting in different binding faces being presented to the analyte. Polyclonal antibodies recognize different epitopes on the same antigen and thus would be considered a heterogeneous ligand. The following equation describes binding of analyte to a heterogeneous ligand:

where B1 and B2 are the two separate binding sites on the ligand and A is the analyte. Note that there are two separate sets of association and dissociation rate constants (ka1/kd1 and ka2/kd2) to describe each binding event. The binding response of a sensorgram from a heterogeneous ligand then, is the sum of the binding response of two separate binding events.
Two-State Conformation Model
The two-state conformation model accounts for the existence of two-conformations of the bound complex. This can happen if binding of the analyte to ligand triggers a change in conformation of the bound complex. The following equation describes the two-state confirmation binding model:
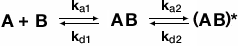
where AB is the first conformation of the bound complex and (AB)* is the second conformation of the bound complex. Once the complex AB forms it can either dissociate to unbound ligand (B) and free analyte (A) or change to the new conformation (AB)*. However, the complex (AB)* must return to the first complex AB before dissociating into unbound ligand and free analyte. The two-state conformation model is very useful for describing an allosteric binding effect where binding of analyte to ligand (a substrate or inhibitor binding to an enzyme, for example) results in a conformational change.
Abdiche YN et al. (2011). Expanding the ProteOn XPR36 biosensor into a 36-ligand array expedites protein interaction analysis. Anal Biochem 411, 139–151.
This article describes in detail how to create a 36 ligand array for antibody screening. This novel use of the ProteOn system enables the immobilization of 36 individual ligands to the array surface. In this assay 36 antibody targets to the same antigen were analyzed using the classical sandwich method and both epitope binning and mapping were performed. All these experiments were achieved on a single ProteOn sensor chip in approximately one day.
Daghestani HN et al. (2010). Theory and applications of surface plasmon resonance, resonant mirror, resonant waveguide grating, and dual polarization interferometry biosensors. Sensors 10, 9630–9646.
This reference describes the theory and application of a number of optical biosensors, including surface plasmon resonance biosensors, and the different configurations for each.
O'Shannessy DJ et al. (1993). Determination of rate and equilibrium binding constants for macromolecular interactions using surface plasmon resonance: use of non-linear least squares analysis methods. Anal Biochem 212,457–468.
This article describes in detail the use of non-linear least squares analysis to fit sensorgram data from the interaction of human sCD4 and a mouse IgG antibody to kinetic equations describing binding with a simple 1:1 binding model (Langmuir model). The authors note that dissociation data should be analyzed first to find a proper dissociation rate constant (kd) so as to calculate a more accurate association rate constant from the association phase data. Using non-linear least squares analysis, the authors demonstrated that the obtained ka and kd values were accurate, as they were independent of the concentration of analyte used in a given SPR experiment.
Protein interaction analysis: analyte interaction and kinetics. Bio-Rad Bulletin 6044.
This Bio-Rad bulletin describes the analysis of SPR data with a simple Langmuir model of 1:1 binding, equilibrium binding, or a Langmuir model with mass transport limitations. Derivation of rate equations from the simple Langmuir model are given, along with integrated versions of these equations, in an effort to describe how data is analyzed using the ProteOn system.
Yousef M (2007). Advances in rapid monoclonal antibody screening. Am biotechnol lab 25, 26–28.
Videos
Learn how the ProteOn can be applied to many important applications of the drug discovery workflow.
Learn how the ProteOn system's design provides data you can trust through unique referencing options.
Learn in more detail how the ProteOn system uses SPR technology to address important experimental questions.

TEST
General ProteOn Literature
| 6411 | ProteOn XPR36 Quantikinetics: Antibody Concentration and Detailed Kinetic Analysis in a Single Experimental Cycle | Click to download |
| 5390 | ProteOn — PIA XPR36 Brochure | Click to download |
| 5538 | ProteOn XPR36 — Analyzing Protein Interaction Array System Featured Article Reprint | Click to download |
| 5413 | ProteOn XPR36 Hardware — 36 Interactions on a Single Chip: Label-Free, in Real-Time PIS | Click to download |
| 5627 | ProteOn Manager Software PIS | Click to download |
| 5404 | ProteOn Sensor Chips — Application-Specific Surface Chemistries = Optimized Ligand Activty PIS | Click to download |
| 5410 | ProteOn Protocol Development Kits PIS | Click to download |
| 5409 | Protein Interaction Analysis — ProteOn XPR36 System Ordering Information Sheet | Click to download |
| 6414 | ProteOn XPR36 Experimental Design and Application Guide, Rev B | Click to download |
Large Molecule Protein-Interaction Analysis
| 3172 | ProteOn PIA — Rapid and Efficient Determination of Kinetic Rate Constants using the ProteOn XPR36 Protein Interaction Array System Tech Note | Click to download |
| 5412 | ProteOn App Guide — Antibody Characterization and Development Using the ProteOn XPR36 PIA System App Guide PIS | Click to download |
| 5540 | ProteOn — PIA Screening, Ranking, and Epitope Mapping of Anti-Human IL-9 Supernatants TN | Click to download |
| 5368 | ProteOn PIA — Analysis of Multiple Protein-Protein Interactions Using the ProteOn XPR Protein Interaction Array System Tech Note | Click to download |
| 5449 | Protein Interaction Analysis Applications of the ProteOn NLC Sensor Chip: Antibody-Antigen, DNA-Protein-Protein Interactions Tech Note | Click to download |
| 5358 | ProteOn PIA — Mechanisms of Protein Binding: Double-Mutant Cycle Analysis Using the ProteOn XPR36 System Tech Note | Click to download |
| 5820 | ProteOn — Rapid Screening and Selection of Optimal Antibody Capturing Agents Using the ProteOn XPR36 Protein Interaction Array System | Click to download |
| 5360 | ProteOn PIA — Rapid and Detailed Analysis of Muliple Antigen-Antibody Pairs Using the ProteOn XPR36 Protein Interaction Array System Tech Note | Click to download |
| 5367 | ProteOn PIA — Rapid Optimization of Immobilization and Binding Conditions for Kinetic Analysis of Protein-Protein Interactions using the ProteOn XPR36 PI Array System Tech Note | Click to download |
Small Molecule Analysis
| 5965 | Rapid High-Throughput Screening of Protein Kinase Inhibitors Using the ProteOn PIA System | Click to download |
| 5797 | Protein Interaction — Rapid Assay Development and Optimization for Small Molecule Drug Discovery Tech Note | Click to download |
| 5679 | ProteOn — Applications of the ProteOn GLH Sensor Chip: Interaction Between Proteins and Small Molecules | Click to download |
| 5960 | High-Throughput Profiling of Kinase Inhibitors Selectivity Using the ProteOn XPR36 Protein Interaction Array System | Click to download |
| 5846 | Determining the Binding Kinetics of HIV-1 Nucleocapsid Protein to Six Densities of Oligonucleotide Using the ProteOn XPR36 Protein Interaction Array System TN | Click to download |
| 5822 | How to Perform Excluded Volume Correction on the ProteOn XPR36 Protein Interaction System PG | Click to download |