
The search for unknown mutations in genomic DNA is important for a broad spectrum of research studies, including fundamental research on gene structure and function, the study of genetic diseases and disorders, and species identification. In any living cell, mutations occur when there is a failure in DNA repair. In this section we review the different types of mutations and some of the techniques that can be used to detect mutations.
Related Topics: Gene Expression Analysis, Gene Cloning and Analysis, and Epigenetics and Chromatin Structure.
Page Contents
Several different types of mutations have been identified:
- Single Base Substitution (aka point mutation or single nucleotide polymorphism [SNP])
A base is exchanged for another (for example, C to T) in the DNA sequence. Depending on the substitution, this type of mutation can change the encoded amino acid (missense mutation) to produce a different protein or an incomplete protein (nonsense mutation) which can lead to a diseased state. An example of this type of mutation is sickle cell disease. - Insertions and Deletions
Single or multiple base-pairs are incorporated or deleted from a DNA sequence. This type of mutation can create frameshifts that cause the mRNA sequence to be not read properly by the translational machinery. Frameshifts can have devastating consequences. An example of this type of mutation is Huntington disease. - Duplications
A section of the genome is doubled. This type of mutation can create overexpression. An example of this type of mutation is high blood pressure. - Translocations
A piece of one chromosome is transferred to a nonhomologous chromosome. An example of this type of mutation is Burkitt lymphoma.
These mutations can be beneficial (or detrimental) depending on which gene they affect. Mutational analysis assists with identifying unknown mutations as well as identifying precursors to diseases and disorders by allowing researchers to study these mutations in a control environment.
Over the past couple of decades, mutation detection techniques, such as denaturing gradient gel electrophoresis (DGGE), constant denaturing gel electrophoresis (CDGE), temporal temperature gradient gel electrophoresis (TTGE), single-strand conformation polymorphism (SSCP), and protein truncation test (PTT), have assisted researchers with analyzing mutations. More recently, high resolution melt (HRM) analysis has also become a technique of choice for mutation detection. All of these techniques require DNA amplification via PCR before the technique can be used. Typically, site-directed mutagenesis is used to create mutant control samples by introducing mutations into some DNA fragments. DNA sequencing can be used to verify the mutation after any of these techniques.
Denaturing gradient gel electrophoresis (DGGE) is an electrophoretic method to identify single base changes in a segment of DNA. Separation techniques on which DGGE is based were first described by Fischer and Lerman. In a denaturing gradient acrylamide gel, double-stranded DNA is subjected to an increasing denaturing environment and will melt in discrete segments called "melting domains." The melting temperature (Tm) of these domains is sequence specific. When the Tm of the lowest melting domain is reached, the DNA will become partially melted, creating branched molecules. Partial melting of the DNA reduces its mobility in a polyacrylamide gel. Since the Tm of a particular melting domain is sequence specific, the presence of a mutation will alter the melting profile of that DNA when compared to wild type. DNA containing mutations will encounter mobility shifts at different positions in the gel than the wild type. If the fragment completely denatures, then migration again becomes a function of size.

In DGGE, the denaturing environment is created by a combination of uniform temperature, typically between 50 and 65°C, and a linear denaturing gradient formed with urea and formamide. A solution of 100% chemical denaturant consists of 7 M urea and 40% formamide. The denaturing gradient may be formed perpendicular or parallel to the direction of electrophoresis. A perpendicular gradient gel, in which the gradient is perpendicular to the electric field, typically uses a broad denaturing gradient range, such as 0–100% or 20–70%.
In parallel DGGE, the denaturing gradient is parallel to the electric field, and the range of denaturant is narrowed to allow better separation of fragments. Examples of perpendicular and parallel denaturing gradient gels with homoduplex and heteroduplex fragments are shown below.
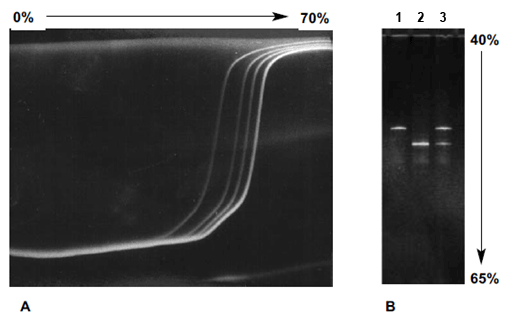
A. Perpendicular denaturing gradent gel in which the denaturing gradient is perpendicular to the electrophoresis direction. Mutant and wild-type alleles of exon 6 from the TP53 gene amplified from primary breast carcinomas and separated by perpendicular DGGE (0–70% denaturant) run at 80 V for 2 hr at 56°C. The first two bands on the left are heteroduplexes and the other two bands are the homoduplexes. B. Parallel denaturing gradient gel in which the denaturing gradient is parallel to the electrophoresis direction. Mutant and wild-type alleles of exon 8 from the p53 gene separated by an 8% acrylamide:bis (37.5:1) gel with a parallel gradient of 40–65% denaturant. The gel was run at 150 V for 2.5 hrs at 60°C in 1x TAE buffer. Lane 1 contains the mutant fragment; lane 2 contains the wild-type fragment; lane 3 contains both the mutant and wild-type fragments.
Constant denaturing gel electrophoresis (CDGE) is a modification of DGGE. In CDGE, the denaturant concentration that gives optimal resolution from a parallel or perpendicular DGGE gel is held constant. The optimal concentration of denaturant to use for a CDGE is determined from the maximum split between wild-type and mutant DNA, as seen in the perpendicular or parallel denaturing gel.
After a mutation has been identified by previous DGGE gels, a CDGE gel can be used to rapidly screen samples for the presence of a mutation. With no gradient required, rapid, high-throughput screening is possible. As in DGGE, the formation of heteroduplex analysis can help in resolving wild-type and mutated fragments when it is not possible to detect a mutation by running homoduplex fragments. An example of a CDGE gel is shown below.

Example of perpendicular DGGE gel used for determining the optimum denaturant concentration used in a CDGE gel. The distance along the gradient where the maximum split seen between samples is 5 cm. The denaturant concentration of the gradient at this distance is 51%. Therefore, the CDGE gel should use a denaturant concentration of 51%.
Temporal Temperature Gradient Gel Electrophoresis (TTGE) exploits the principle on which DGGE is based, without requiring a chemical denaturing gradient. Amplified mutant and wild-type DNA from the gene of interest is loaded onto a polyacrylamide gel containing a constant concentration of urea. During electrophoresis, the temperature is increased gradually and uniformly. The result is a linear temperature gradient over the length of the electrophoresis run. Thus, a denaturing environment is formed by the constant concentration of urea in the gel in combination with the temporal temperature gradient. With no chemical gradient required, rapid, high-throughput screening is possible.
Temporal temperature gradient gel. Amplified mutant and wild-type allelss of exon 7 from the cystic fibrosis gene. Separation by TTGE run at 130 V for 5 hours in 1.25x TAE buffer on a 6 M urea/6% acrylamide gel (37.5:1) using a temperature range of 50–60°C and a ramp rate of 2°C/hr. Lane 1, mutant allele (1154 insTC); lane 2, mutant allele (G330X); lane 3, mutant allele (deltaF311); lane 4, mutant allele (R334W); and lane 5, wild-type allele.
Single-strand conformation polymorphism (SSCP) technique is based on the fact that single-stranded DNA has a sequence-specific secondary structure. Sequence differences as small as a single base change can affect this secondary structure and can be detected by electrophoresis in a nondenaturing polyacrylamide gel. Double-stranded mutant and wild-type samples are first denatured into single strands and then loaded onto the gel. Differences in mobility of the single strands between the control wild-type DNA and the other samples indicate a mutation. SSCP is a widely used mutation screening method because of its simplicity. However, since experimental conditions cannot be predicted for a particular DNA, it is important to optimize gel electrophoresis conditions. The ability to detect single base changes rests on several factors which optimize band resolution.
- Fragment size — the estimated efficiency for detecting single base changes is 90–95% for fragments less than 350 bp, but the efficiency will decrease as the length of fragment increases.
- Gel temperature — migration differences due to a single mutation are observed at buffer temperatures between 4–25°C. Optimal temperature must be determined empirically.
- Gel additives — in some cases, 5–10% glycerol can be added to the gel to improve the mobility differences in fragments. Since glycerol can reduce the mobility of single-stranded DNA fragments at low temperatures, it is typically used with gels run near room temperature.
- Crosslinking ratio — the acrylamide/bis ratio determines the percent of crosslinking. SSCP gels generally use 1–2 % crosslinking. Acrylamide concentrations will vary from 5 to 10%.
- Buffer concentration — gels are run with TBE buffer at concentrations of 0.5x or 1.0x. In some cases, 0.5x TBE appears to give slightly better results than 1.0x TBE.
SSCP protocols have typically used radioisotope-labeled fragments, but recently nonradioactive or "cold SSCP" methods have been developed.
Protein Truncation Test (PTT) is a mutation screening method that detects truncated proteins after translation of the coding sequence. There are six steps associated with the PTT assay. The first step is to amplify by PCR a template RNA sample. The second step requires a reverse transcriptase reaction of the starting mRNA to make cDNA. The third step amplifies the sequence of interest and incorporates a tailed primer sequence. This tailed primer contains a T7 promoter and eukaryotic translation initiation sequence. These sequences are needed for in vitro transcription and translation. The fourth step checks the PCR product on an agarose gel for quality, size, and approximate quantity. In the fifth step, the PCR products are transcribed with RNA polymerase and translated into peptides. There are commercial kits that couple the transcription and translation reaction in one tube using rabbit reticulocyte lysate. Detection of the translation products is done by adding radiolabeled amino acids, typically 3H-leucine or 35S-methionine, to the translation reaction. The final step involves analyzing the translation products on an SDS-PAGE gel to determine their length. The gel is normally treated with a fluorographic-enhancing reagent to reduce the exposure time to X-ray film. Truncated proteins are identified by size differences when compared to full-length control proteins.
High Resolution Melt (HRM) analysis uses DNA melt curve profiles that are both specific and sensitive enough to distinguish nucleic acid species based on small sequence differences for mutation scanning, methylation analysis, and genotyping. The use of HRM for mutation scanning is becoming a more popular technique for high-throughput environments because HRM:
- Decreases the risk of contamination
- Increases throughput
- Saves on cost and turnaround time
Cheng J et al. (2011). Detection of hemi/homozygotes through heteroduplex formation in high-resolution melting analysis. Anal Biochem 410, 158–160. PMID: 21111703
Cocolin L et al. (2001). Denaturing gradient gel electrophoresis analysis of the 16S rRNA gene V1 region to monitor dynamic changes in the bacterial population during fermentation of Italian sausages. Appl Environ Microbiol 67, 5113–5121. PMID: 11679334
Gorre ME et al. (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880. PMID: 11423618
Sørlie T et al. (2005). Mutation screening of the TP53 gene by temporal temperature gradient gel electrophoresis. Methods Mol Biol 291, 207–216. PMID: 15502225
Teixeira MR et al. (2000). High frequency of clonal chromosome abnormalities in prostatic neoplasms sampled by prostatectomy or ultrasound-guided needle biopsy. Genes Chromosomes Cancer 28, 211–219. PMID:10825006
Temesvári M et al. (2011). High-resolution melting curve analysis to establish CYP2C19*2 single nucleotide polymorphism: Comparison with hydrolysis SNP analysis. Mol Cell Probes 25, 130–133. PMID: 21315147
Wang Y et al. (2003). Novel identification of a four-base-pair deletion mutation in PITX2 in a Rieger syndrome family. J Dent Res 82, 1008–1012. PMID: 14630904