

Join Us for In-Depth Technical Courses in Real-Time qPCR
Become a qPCR expert through our free 5-part webinar series.
On This Page |
Identification of Genomic Targets | Quantification of Genomic Targets | Understanding Gene Function | Gene Expression | Real-Time qPCR Learning Center | Related Topics & Products |
The Genomics Learning Center is a knowledge hub where you can find both general information on a range of genomics technologies and advanced solutions for PCR, qPCR/RT-PCR, digital PCR, and next-generation sequencing (NGS). The field of genomics research has grown incredibly and is constantly evolving, with new technologies pushing the boundaries of the study of genes and gene function. Whether you are an experienced genomics researcher or new to the field, it is critical to stay informed about the latest applications and techniques so you can design your experiments with confidence and drive your research forward.
Identification of Genomic Targets
Next-generation sequencing (NGS) and advanced PCR methods have revolutionized genomics and made sequencing accessible to a wide range of research applications. New technologies continue to advance long-range, direct, and single-cell sequencing, while new library preparation and nucleic acid quantification solutions help optimize workflows. Topics here include standard and emerging NGS methodologies for sequencing DNA and RNA targets plus advanced digital PCR tools for a range of genomics applications.
Latest Methods for the Identification of Genomic Targets
-

DNA Challenges:
Mining the Genome
-

DNA Solutions:
Advances in NGS Technologies
-

RNA Challenges:
Understanding a Complex Transcriptome
-

RNA Solutions:
Advances in RNA-Seq Technologies
Quantification of Genomic Targets
PCR technology remains vital to genomic workflows, with new high-performance digital PCR systems offering vast improvements in accuracy and reproducibility. Digital PCR systems like droplet digital PCR (ddPCR) allow ultrasensitive and absolute nucleic acid quantification. While real-time PCR (qPCR) is often used for target detection, these droplet digital PCR systems provide exceptional sensitivity in detection of low abundant targets or complex targets, allelic variants (SNPs), or subtle changes in target levels. Topics here include the application of digital PCR technology to gene expression analysis, CNV analysis, and single-cell studies.
Latest Methods for the Quantification of Genomic Targets
-

DNA Challenges:
Searching for Rare Events
-

DNA Solutions:
ddPCR: Finding Needles In Haystacks
-

RNA Challenges:
Quantifying Gene Expression
-

RNA Solutions:
Tools for Tracking Expression Patterns
Understanding Gene Function
The ultimate goal of genomics is to understand the function of genes in the context of health and disease. This is typically achieved by mutating or omitting the gene in question and analyzing the downstream cellular effects. CRISPR technology has greatly expanded our tool kit for targeted gene editing and introduced opportunities for novel gene therapies. Advancements in NGS are also allowing scientists to explore more of the non-coding transcriptome and revealing the diverse roles of non-coding RNAs. This section will explore topics in functional genomics, including genome editing, non-coding RNAs, and RNA-Seq data analysis.
Latest Methods for Understanding Genomic Function
-

DNA Challenges:
Mutation or Polymorphism
-

DNA Solutions:
CRISPR and Synthetic Biology
-

RNA Challenges:
Coding or Non-Coding?
-

RNA Solutions:
Tools for Tracking Expression Patterns
More on Gene Expression
-

Challenges and Solutions in Gene Expression Research
Learn to identify and tackle the important, yet surmountable, challenges faced by modern gene expression studies to set your lab up for success.
-

How Different Molecular Techniques Synergize in Gene Expression
Implementing a fit-for-purpose combination of gene expression analysis techniques can yield better outcomes, save you time and money, and help you make discoveries faster.
-

Gene Expression: Where Has It Been, and Where Is It Going?
Astounding technological developments have drastically improved the sensitivity, throughput, and spatial analysis capabilities of gene expression research over the decades. Where will it go next?
-

Genetic Profiling Paving Ways to Oncology Insights
Oncology research is the foundation of the fight against cancer. Here's how genetic profiling can improve early detection, monitoring, and the development of novel therapeutics for various cancers.
-

Tips for New Labs Incorporating Gene Expression
Setting up a new research lab can be a daunting task, but it doesn't have to be overwhelming. By considering three key factors up front, investigators can establish their gene expression research pipelines with minimal issues.
-

Important Considerations in Gene Expression Studies
Modern gene expression studies face several important, yet surmountable, challenges. Address these factors head on by knowing them beforehand.
-

Your Guide to MIQE and dMIQE
In this article, we summarize MIQE and dMIQE guidelines and explore implications and benefits for standardizing qPCR and ddPCR™ experiments.
-

Balancing qPCR and ddPCR™ in Therapeutic Development
Gene expression analysis is a critical part of the drug discovery and development process. Here's how RT-qPCR and ddPCR can be used to support various stages across the drug development pipeline, from discovery to manufacture.
-

Benefits of Complementary Methods in Gene Expression Profiling
Drug development isn't a one-size-fits-all process, so the tools you use in your research shouldn't be either. Combining multiple powerful gene expression analysis techniques can bring your drug development efforts to the next level.
-
A Perfect Match for Your Gene Expression Research Needs
Combine research tools to create a robust gene expression analysis platform that can address every changing need posed by your research.
-

The Role of qPCR and ddPCR™ in Potency Analysis
Learn about the multiple advanced nucleic acid quantification technologies that can support accurate, streamlined potency assays.
-

Leveraging Molecular Technologies to Stay on Top of the Scientific Game
Learn about using functional genomic techniques to find commonalities among diverse tumor cells.
-

The Groundbreaking mRNA Therapeutics Revolution
Whether quantifying an mRNA-based drug, measuring therapeutic effects on mRNA levels, or detecting rare contaminants, Droplet Digital™ PCR is transforming mRNA therapeutics.
-

Tips and Tricks for Optimum Assay Design
When deciding whether a qPCR assay or a ddPCR™ assay will best meet your needs, it's important to carefully consider the specific research question you're seeking to answer.
-
Selecting Your Next PCR System?
This PCR selection infographic can help you determine which PCR solution is best suited for your research needs.

Related Topics and Products
-

PCR (Polymerase Chain Reaction)
Learn how PCR works, how to choose the right PCR instrument, how to design, optimize, analyze, and troubleshoot PCR assays.
-

Real-Time PCR (qPCR)
Discover an array of solutions to optimize, execute, and troubleshoot MIQE-compliant real-time PCR experiments.
-

Digital PCR (ddPCR)
Learn how breakthrough digital PCR technology is used in droplet digital PCR (ddPCR) to provide ultrasensitive and absolute nucleic acid quantitation.
-
SARS-CoV-2 / COVID-19 Assay and Research Solutions
Bio-Rad provides a wide range of products for use in the support of COVID-19 diagnostic screening, confirmation of test results, surveillance, and therapeutic & vaccine research and development.
-

ddPCR Master Class Videos
Become an expert in all things ddPCR. Our master class sessions allow you to learn from the best in the field. From instrument overviews to informational sessions highlighting the implications of ddPCR in the real world, Master Classes have it all.
-

Targeted Genome Editing
Learn about the major systems for targeted genome editing, including CRISPR/Cas9, some key applications, and workflow options.
-
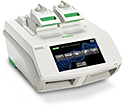
Traditional PCR Systems
DNA amplification instruments range from personal thermal cyclers to the flexible 1000-series.
-

qPCR Detection Systems
These systems deliver sensitive, reliable detection of both singleplex and multiplex real-time PCR reactions.
-

Digital PCR
Bio-Rad's unique Droplet Digital PCR technology provides absolute quantification of nucleic acids for a wide range of applications including cancer mutation studies, HIV quantification, and environmental monitoring.
-

PCR Plastic Consumables
A large selection of thin-wall PCR tubes, plates, seals, and accessories precisely manufactured for optimal fit and cycling performance in Bio-Rad thermal cyclers, real-time PCR systems, ddPCR systems, and all major competitor systems.
-

PCR & qPCR Reagents
A wide range of reagents for reverse transcription, PCR, and real-time PCR, optimized to generate accurate and reproducible data.
-
Next-Generation Sequencing
To assist researchers in answering the most complex biological questions, Bio-Rad offers a number of novel products for using NGS library preparation to enable cutting edge technologies, such as whole transcriptome sequencing (WTS) and single-cell sequencing.
-

PrimePCR PCR Primers, Assays & Arrays
Experimentally validated PCR primer and probe assays for gene expression, copy number variation, and mutation detection analysis for real-time PCR and Droplet Digital PCR.
-
Droplet Digital PCR Assays
Bio-Rad offers a comprehensive portfolio of Digital PCR Assays and Kits for numerous applications including Mutation Detection, Copy Number Determination, Genome Edit Detection, Gene Expression, Expert Design Assays, Residual DNA Quantification and Library Quantification.

COVID-19 Variant Detection with RT-qPCR Poster
2022 European Congress of Clinical Microbiology & Infectious Diseases (ECCMID) Conference
Learn how UCSF used PrimePCR SARS-CoV-2 single and multiple mutation assays to detect 4 SARS-CoV-2 variant mutations in a single well, on samples with Cq<40.