
In recent years, drug discovery researchers have expanded their portfolio of products from small-molecule drugs to large biomolecules such as recombinant proteins or monoclonal antibodies developed for diagnostic or therapeutic uses. The production of these important biomolecules often requires many chromatographic steps to obtain pure products and ensure that no contaminants are present that might cause adverse effects such as allergic reactions or even death.
Related Topics: Histidine-Tagged Recombinant Protein Purification and On-Column Refolding, Affinity Chromatography, and Protein Purification and Isolation.
Page Contents
The amount of purified protein required differs depending on the research and development phase in which the biomolecule of interest is being evaluated. For example, in the early discovery phase, smaller amounts of multiple antibody constructs, each with different binding characteristics, are required for testing and analysis.
Methods such as surface plasmon resonance are used to determine the kinetic parameters for the binding of these antibodies with their targets and to allow for the identification of antibodies that have the desired binding characteristics. These antibodies are then further investigated in the preclinical phase. Ultimately, clinical trials are performed to determine the effectiveness of experimental drugs in humans.
Once an effective antibody has been identified, the last phase of the drug-production workflow requires large amounts of purified product. At the large scale, similar separation techniques are utilized but with a much higher throughput.
There are many different methods for antibody production; two of the most popular methods are the creation of antibodies and antibody fragments from mouse hybridoma cell lines, and phage display. Phage display is a common method for the production of antibody fragments (Fabs), so named because these fragments contain only an antigen-binding region (see Figure 1).
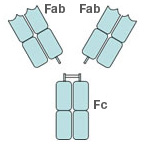
Fig. 1. Schematic structure of an antibody. Fab, antigen binding region; Fc, crystallizable region.
Phage display is widely used by researchers in the development of novel therapies or the identification of novel targets for therapies because millions of Fabs can be produced in a single experiment. These Fabs are subjected to an initial screening to identify those with highest affinity for their intended targets. Successfully selected Fabs are often converted into full-length antibodies for possible therapeutic use; however, successful translation into clinical practice remains rare.
Both methods rely on chromatography to isolate and purify antibodies and antibody fragments (Fabs) from complex media, such as cultured mouse cell lines or bacterial supernatants. Because these media contain highly complex protein mixtures, extensive multistep chromatography is often required to produce purified antibody products.
Monoclonal antibodies are typically purified from crude samples derived from either culture media or ascites taken from a host animal. These crude samples contain many contaminants such as growth factors, hormones, DNA, endotoxins, and host cell proteins.
A typical workflow for monoclonal antibody purification involves initial centrifugation and filtration steps prior to chromatography. Many chromatographic methods can be employed in antibody purification.
A popular method utilizes affinity chromatography. Here, the crude sample is passed through a column filled with a resinous stationary phase containing protein A, which captures the antibody because protein A has a high specificity for the Fc (fragment, crystallizable) regions of antibodies (see Figure 1).
Although affinity chromatography is a simple and quick approach, it has the drawback of being expensive compared to other methods. Much of this expense is due to the shorter lifetime of protein A resin compared with other stationary phases.
Additionally, affinity chromatography is only a part of a multistep approach wherein the crude sample is first passed through an affinity column and subsequently polished using ion exchange chromatography (IEX).
Cation exchange chromatography is a popular method for cleaning up an isolated monoclonal antibody. In this step, the antibody binds the solid phase, and contaminants are allowed to flow through or wash off the protein.
The complementary method, anion exchange chromatography, may also be employed, in which case contaminants are captured on the anion exchange column, while the antibody flows through. Two ion-exchange chromatography steps are often needed for the complete removal of cell-related contaminants.
Size-exclusion chromatography (SEC) is a less popular final step for removing residual proteins and contaminants; this method often employs a size-based filter rather than a column due to volume constraints.
Tagged affinity chromatography is another frequently employed method. In this process, a recombinant antibody is produced as a fusion protein containing a terminal affinity tag such as a polyhistidine tag. The chromatography column contains media functionalized with a molecule that binds the tag with high affinity; for example, transition metal compounds, especially nickel compounds such as tris-NTA, bind histidine residues tightly yet reversibly. This method enables antibody capture and purification in one step in a process called immobilized metal ion affinity chromatography (IMAC). However, this approach yields an antibody with an attached tag, which may interfere with the binding of the antibody to its target antigen. This tag must therefore be removed, adding downstream processing to the workflow.
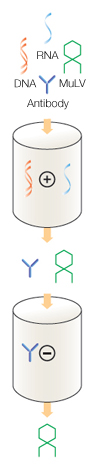
Fig. 2. Chromatography process flow diagram for antibody purification and virus removal.
Therapeutic antibody production on the process scale demands tens of grams of highly pure product. This product must be free from contaminants such as viruses that may cause adverse events in patients. The production of virus-free cetuximab provides an illustrative example.
Cetuximab is a chimeric monoclonal antibody with a high specificity for human epidermal growth factor receptor. This antibody is currently used to treat patients with metastatic cancers.
Several different methods have been tested to produce a virus-free antibody product; one such method using a two-column chromatographic procedure is described below.
In this scaled-down example, the antibody is grown in serum-free production media and spiked with the Moloney ecotropic murine leukemia virus (MuLV). This model envelope retrovirus is approximately 80–110 nm in diameter.
The purification process involves a two-column chromatographic procedure utilizing strong anion and strong cation exchange chromatography (shown to the side). A known titer of the MuLV, here 1.9 × 106 plaque-forming units (PFU) per ml, is spiked into the antibody preparation and loaded onto an anionic column.
This column strongly binds contaminants such as DNA and RNA, and the effluent contains purified protein and MuLV at a lower concentration than in the original solution (5.75 × 105 PFU/ml).
The effluent is then loaded onto the cation exchange column, where the antibody binds to the column. The column is washed free of contaminants, and the antibody is then eluted by applying a pH gradient.
This two-column method successfully yields purified antibody and reduces viral load eightfold, thus meeting the U.S. Food and Drug Administration safety requirements.