
On This Page |
How Real-Time PCR Works | Top 3 Hallmarks of an Optimized RT-qPCR Assay | Related Products | Resources |
Once RNA is transcribed into DNA, RT-qPCR occurs under the catalytic activity of a specific DNA polymerase that is only able to amplify fragments from DNA molecules. In RT-qPCR, the amplification reaction is set up with PCR reagents, primers and probes in a master mix, and run using real-time PCR instruments.
The RT-qPCR assay is based on the principle that a higher or lower initial amount of a specific DNA sequence will lead to higher or lower concentrations of amplicons for that DNA sequence. During amplification, the PCR protocol will use a fluorescence dye to tag the newly synthesized sequences and allow their detection. The target-specific fluorescence from the amplification reaction is monitored in real-time and is proportional to the amount of amplified product.
Real-time PCR results can either be qualitative (presence or absence of a sequence) or quantitative (number of copies of DNA). Real-time PCR that is quantitative is known as qPCR. The sections below provide an overview of the amplification process which is central to qPCR.
How Real-Time PCR Amplification Works
To understand how real-time PCR works, we illustrate a qPCR analysis using a typical amplification plot (Figure 1). In this plot, the number of PCR cycles is shown on the x-axis, and the fluorescence from the amplification reaction, which is proportional to the amount of amplified product in the tube, is shown on the y-axis.
The amplification plot shows two phases, an exponential phase followed by a non-exponential plateau phase. During the exponential phase, the amount of PCR product approximately doubles in each cycle. As the reaction proceeds, however, reaction components are consumed, and ultimately one or more of the components becomes limiting. At this point, the reaction slows and enters the plateau phase (cycles 28–40 in Figure 1).
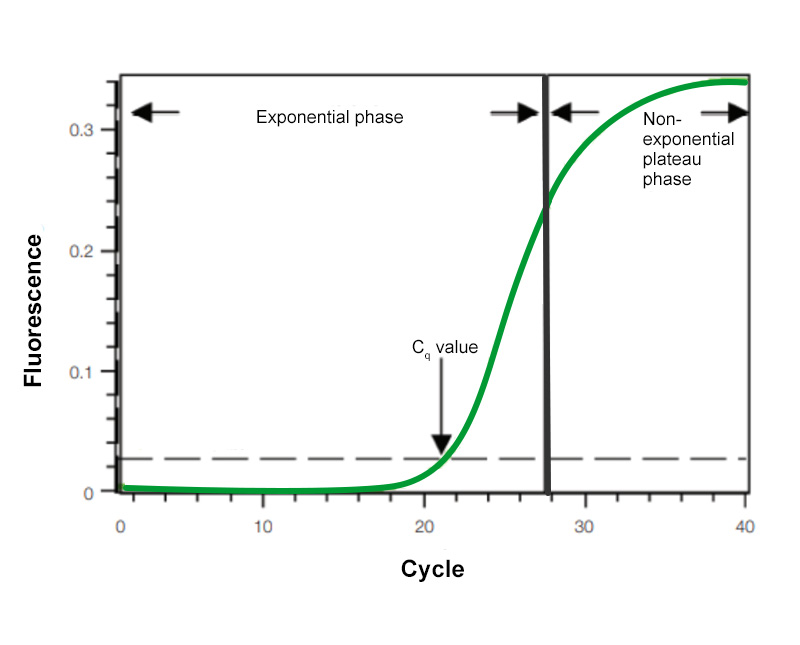
Figure 1. Amplification plot. Baseline-subtracted fluorescence versus number of PCR cycles.
Top 3 Hallmarks of an Optimized qPCR Assay
Since real-time quantification is based on the relationship between initial template amount and the CT value obtained during amplification, an optimal qPCR assay is absolutely essential for accurate and reproducible quantification of your sample. The hallmarks of an optimized qPCR assay are:
- Linear standard curve (R2 > 0.980 or r > |–0.990|)
- High amplification efficiency (90–105%)
- Consistency across replicate reactions
A powerful way to determine whether you qPCR assay is optimized to run serial dilutions of a template and use the results to generate a standard curve. The template used for this purpose can be a target with known concentration (e.g., nanograms of genomic DNA or copies of plasmid DNA) or a sample of unknown quantity (e.g., cDNA). The standard curve is constructed by plotting the log of the starting quantity of template (or the dilution factor, for unknown quantities) against CT value obtained during amplification of each dilution. The equation of the linear regression line, along with Pearson's correlation coefficient (r) or the coefficient of determination (R2), can then be used to evaluate whether your qPCR assay is optimized.
Learn More about the Hallmarks of an Optimized qPCR Assay »
Assay Components in a qPCR Protocol
In multiplex qPCR, multiple targets are amplified in a single reaction tube. Each target is amplified by a different set of primers and a uniquely-labeled probe that will distinguish each PCR amplicon. Multiplexing provides some advantages over single-reaction PCR, including requiring an overall lower amount of starting material, increased throughput, lowered reagent costs, and less sample handling. However, the experimental design for multiplexing is more complicated because the amplification of each target can affect others in the same reaction. Therefore, careful consideration of design and optimization of the reactions is critical. To incorporate all necessary parameters, we strongly recommend using a design tool for primers and probes.
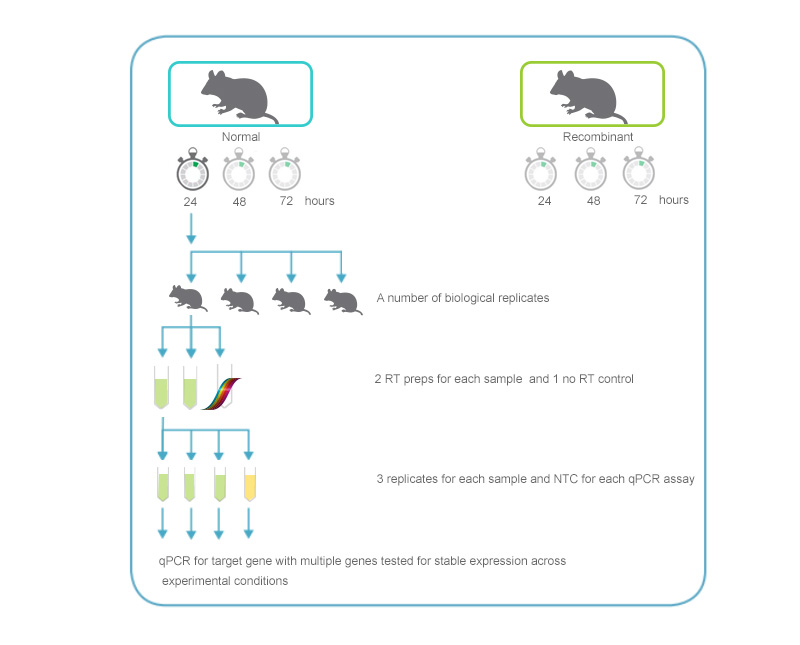
Figure 2. qPCR assay setup. An overview of the replicates and controls required for an experiment comparing 2 different samples analyzed at several time points after treatment. The illustration includes 3–5 biological replicates for each time point in the experiment. For each replicate, 2 reverse transcription reactions (+RT) and 1 with no RT (-RT control) will be performed. For each cDNA sample generated, 3 technical replicates for qPCR analysis will be set up. The example also includes a "no-template control" for each gene analyzed to identify any signal due to contamination.
Reaction Components for a qPCR Assay Protocol
Template
qPCR requires a few copies of a target nucleic acid (equivalent to about 100 pg of gDNA or cDNA) to initiate the reaction. To minimize contamination with reaction inhibitors, the starting template amount should be kept to the minimum required to achieve accurate quantification. If starting with RNA, primer design and DNase I treatment can reduce background signals generated from gDNA contamination.
Primers
Whether using a dsDNA-binding dye or a probe-based detection chemistry, designing high-quality primers is one of the most critical pre-experimental steps in qPCR. See the sections on qPCR Assay Development for more information. The melting temperature for your assay should be based on the different lengths of your assay primers, as shown in Figure 3.
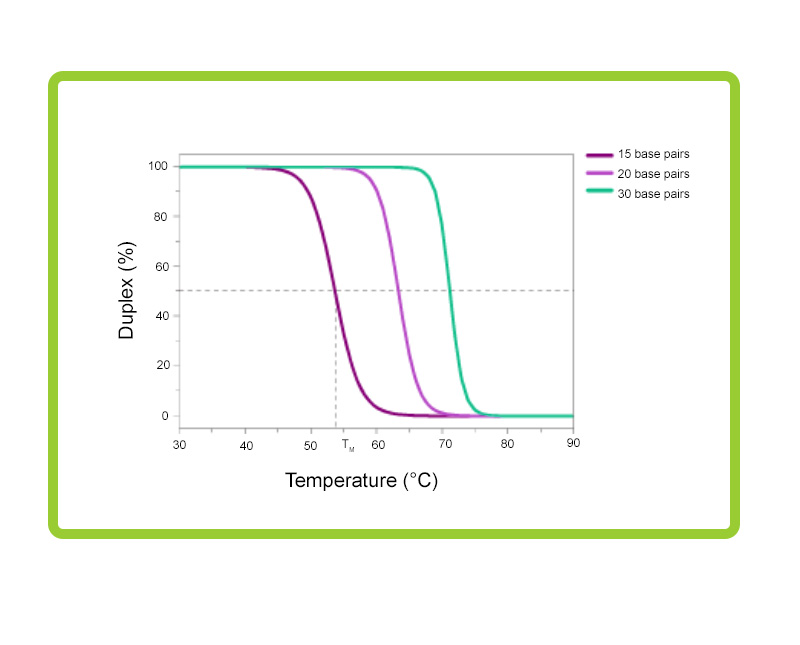
Figure 3. Melting profiles for primers of various lengths. The melting temperature (Tm) of the primers is dependent on their length in base pairs. It is best to keep the Tm of the different primers between 55°C and 75°C, and within 5°C of each other. In the reaction shown here, a PCR buffer was used which contained 1 mM Mg2+ 50 mM KCl, 10 mM Tris. GC content for all primers was ~50%.
Probe(s)
To maximize sensitivity and specificity, the appropriate probe type for the application should be selected. When using probes as the amplicon detection mechanism, primer dimers and nonspecific products should be avoided because they can lower reaction efficiency.
dNTPs
Oligonucleotides need to be included in the amplification reaction mix. Standard qPCR master mixes contain dATP, dCTP, dGTP, and dTTP. However, some master mixes are available that replace dTTP with dUTP. Note that products from previous reactions run with dUTP will contain uracil instead of thymine, and they will be susceptible to cleavage by Uracil-DNA-Glycosylase (UNG). Therefore, prior incubation of subsequent reactions with UNG prevents carryover contamination between reactions. For this procedure to be effective, all PCR reactions carried out in the laboratory must use dUTP.
Magnesium
Magnesium chloride (MgCl2+) is necessary for reverse transcriptase, Taq DNA polymerase and Taq DNA 5’ to 3’ exonuclease activity. Optimal Mg2+ concentrations for reactions are usually between 3–6 mM. While most commercial master mixes contain MgCl2+, it can be necessary to optimize the concentration further. In some cases, a reaction mix that does not contain MgCl2+ may be required so that a low concentration can be used for detection of specific types of probes.
Reverse Transcriptase
A reverse transcriptase enzyme which enables high yields of cDNA, with activity at high temperature, is essential for RT-qPCR. Assay performance at elevated temperatures help to ensure that regions of RNA with significant secondary structure are destabilized and accessible for hybridization and subsequent amplification. When performing one-step RT-qPCR, high-temperature activity allows the use of gene-specific primers with high melting temperatures (Tm), and increases reaction specificity. For two-step protocols, it is important to ensure that the enzyme results in a linear and proportional yield of cDNA from RNA).
Taq DNA Polymerase
Just as the appropriate reverse transcriptase is essential for the RT, selection of the appropriate polymerase enzyme is needed as well. A critical issue of natural Taq DNA polymerase is that the enzyme has residual polymerase activity at low temperature, resulting in nonspecific primer binding and thus, nonspecific product formation. Using antibody-blocked or chemically blocked Taq DNA polymerases (‘hot start’) can help to rectify this situation by preventing enzyme activity until the high-temperature denaturation step begins.
Buffer
Buffers or reaction master mixes, typically contain dNTPs, a Taq DNA polymerase, MgCl2 and stabilizers. Other optional dyes may be included, such as SYBR Green I dDye, ROX™, fluorescein and inert loading dyes (Loading Control Dyes), depending on the detection chemistry, instrumentation, and reaction requirements. Commercial PCR buffer components and stabilizers are typically proprietary. Purchasing the ingredients together as a master mix increases batch consistency and convenience while reducing the number of pipetting steps, and thus, the chance for error and contamination.
Loading Control Dyes
Some real-time PCR thermal cycler systems require a loading dye such as ROX to be included into each reaction to control for optical system variability and to normalize differences in signal intensity. (Bio-Rad real-time thermal cyclers do not require ROX.) Likewise, some thermal cyclers require an initial fluorescein signal to create a virtual background when working with SYBR Green I dDye aAssays (which have very low background). These may be included in the master mix or as separate components.
Assay Controls and No-Template Controls (NTC)
For each assay type, positive controls, negative controls, and no-template controls are essential. Positive controls ensure that the assay can correctly amplify if the target is present, while negative controls help show the negative baseline for the assay. No-template controls help identify sample contamination from user error.
Negative controls
No-template control — water is added in place of the RNA template from a reaction. This control helps identify sample contamination from user error.
A no-reverse–transcriptase control (–RT) omits the reverse transcriptase in the reverse-transcription step of a RT-qPCR. The purpose of this control is to assess the amount of genomic DNA contamination present in an RNA preparation. Perform this control for any assay that might amplify genomic DNA.
A no-amplification control lacks the DNA polymerase used for PCR. It is a control for background fluorescence of the PCR assay and probe stability.
Positive Controls
An exogenous positive control is external DNA or RNA which contains a target of interest. Using this control can help identify any components in the sample which might inhibit the reverse transcription and/or PCR amplification process. (gBlocks Gene fragments, custom genes, and Ultramer DNA Oligonucleotides can serve as exogenous positive controls with known starting copy number.)
An endogenous positive control is a native target found in the experimental sample of interest which offers a normalizer or reference among samples, and often uses a housekeeping gene. These control reactions will correct for quantity and quality differences among samples and ensure accurate internal controls. Choice of an appropriate normalizing gene to use will depend on the RNA source and experimental conditions of the sample. To screen multiple genes under the experimental conditions employed to determine which expression levels fluctuate the least, use programs, such as qBase+ (Biogazelle) to evaluate the performance of various normalizers. If you choose a normalizing gene from the literature, be sure to perform a reaction to verify that the gene expression levels do not fluctuate across samples before you use it as a control.
Thermal Cycling Protocol for PCR Amplification
Below is an example of a thermal cycling protocol for an RT-qPCR assay run on a CFX Opus 96 System
| Step | Description | Temperature °C | Time | Number of Cycles |
|---|---|---|---|---|
| 1 | Reverse transcription | 50 | 10 min | 1 |
| 2 | DNA polymerase activation and template denaturation | 95 | 10 min | 1 |
| 3 | Amplification - Template denaturation | 95 | 10 sec | 45 |
| 4 | Amplification - Annealing/extension and plate read on a CFX System | 58 | 30 sec | 45 |
* Set the lid temperature to 105°C.
Related Products

qPCR Detection Systems
These systems deliver sensitive, reliable detection of both singleplex and multiplex real-time PCR reactions.

PCR Plastic Consumables
A large selection of thin-wall PCR tubes, plates, seals, and accessories precisely manufactured for optimal fit and cycling performance in Bio-Rad thermal cyclers, real-time PCR systems, ddPCR systems, and all major competitor systems.
Resources

End the Cycle of Repeated, Failed Experiments with a Robust Field Guide to qPCR
A snapshot of the critical steps needed to achieve excellent qPCR results.

Tips, Tricks & Best Practices for qPCR
The Ultimate qPCR Guide.

PCR Amplification Guide
A guide for RT-PCR on key concepts, experimental designs, analyses, and product recommendations.

Amplification and Reagents and Plastics Brochure
An overview of PCR reagents and plastics with info on specifications, performance data, and more.
Validating a Quantitative PCR (qPCR) Experiment to Minimize Error and Maximize Data Quality
A checklist of critical steps needed to achieve excellent results to performing a valid qPCR experiment.