
On This Page | History of Transcriptomics | RNA-Seq Technologies | Applications of RNA-Seq | Related Topics and Products |
Every cell in the human body contains the same genomic content, yet differential gene expression leads to highly varied phenotypes at the level of individual cells. A detailed analysis of the transcriptome, which is the total collection of transcribed RNA in an organism – mRNA, rRNA, tRNA, and noncoding RNA – is essential to understanding genomic function as it pertains to both normal biology and diseases like cancer.
RNA sequencing (RNA-Seq) allows scientists to determine the quantity and sequence of RNA in an organism, tissue or single cell, using next-generation sequencing (NGS) methodologies. Thus, the standard RNA sequencing protocol involves the added step of reverse transcription into complementary DNA (cDNA) prior to sequencing on an NGS platform. Developments in NGS have revolutionized the field of transcriptomics and led to protocols for studying differential gene expression, alternate splicing, gene fusions, and post-translational modifications as well as more and to more recent applications in single cells.
This article provides a brief overview of RNA-Seq history, in terms of short-, long-, and direct-read methods, and current applications.
History of Transcriptomics
Early Methods
Starting in the 1970s, scientists used reverse transcriptase to store purified RNA in the more stable cDNA format. In the 1980s and 1990s Frederick Sanger’s dideoxy chain termination method was applied to sequencing the cDNA of random transcripts called expressed sequence tags (ESTs), which was a way to survey genes without whole-genome sequencing. Initially, low-throughput Northern blot and reverse transcriptase quantitative PCR (RT-qPCR) methods were used to quantify transcriptome activity. Later, the serial analysis of gene expression (SAGE) method was developed to analyze cDNA concatemers using Sanger sequencing. These methods were laborious and have been largely replaced by newer techniques.
Modern Technologies
High-density microarrays, introduced in the mid-1990s, represent the first of the so-called contemporary methods for transcription profiling. A DNA microarray, also known as DNA chip, contains hundreds of thousands of microscopic spots of DNA probes on a solid surface that hybridize to specific targets based on sequence complementarity. Imaging of the chip following hybridization to fluorescently labeled sample is then analyzed to determine the abundance of each gene based on the relative intensity at each probe site. Improvements in specificity, sensitivity, and detection made microarrays the method of choice for gene gene-profiling studies until the late 2000s (Lowe 2017).
Next-Generation RNA Sequencing: RNA-Seq
The advent of next-generation sequencing in the early 2000s enabled RNA sequencing in a massively parallel format and without the requirement for prior sequence knowledge. In the standard RNA-Seq workflow, RNA is first isolated from the experimental sample and enriched for the RNA type of interest, e.g., mRNA. This enrichment step is critical because rRNA constitutes 80% of the cell’s RNA content, easily outcompeting the more experimentally significant RNA content, like mRNA and microRNA, in read count. Additionally, various quality-control steps are needed to ensure that the enriched RNA is of sufficient quality and quantity for sequencing. Next, high-quality RNA is fragmented, converted to cDNA, and sequenced on an NGS platform (Fig 1).
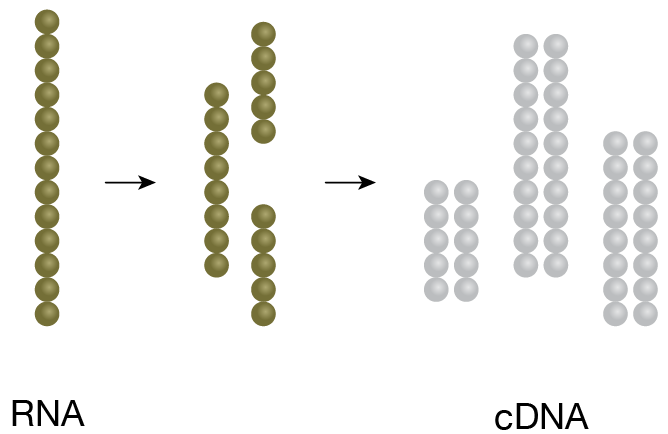
1. cDNA Generation
RNA of interest is fragmented and converted to cDNA.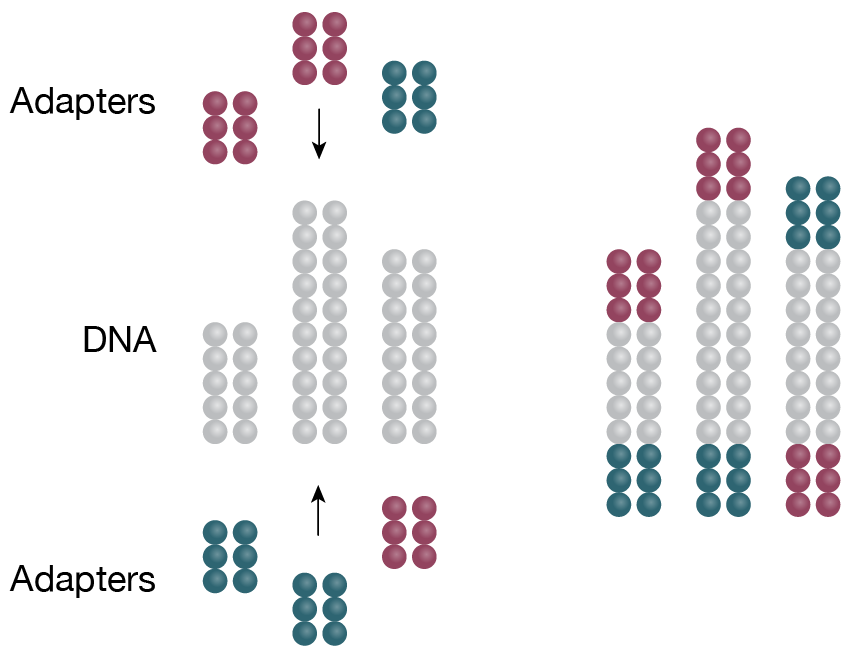
2. Library preparation
cDNA is ligated to specific adaptors in preparation for amplification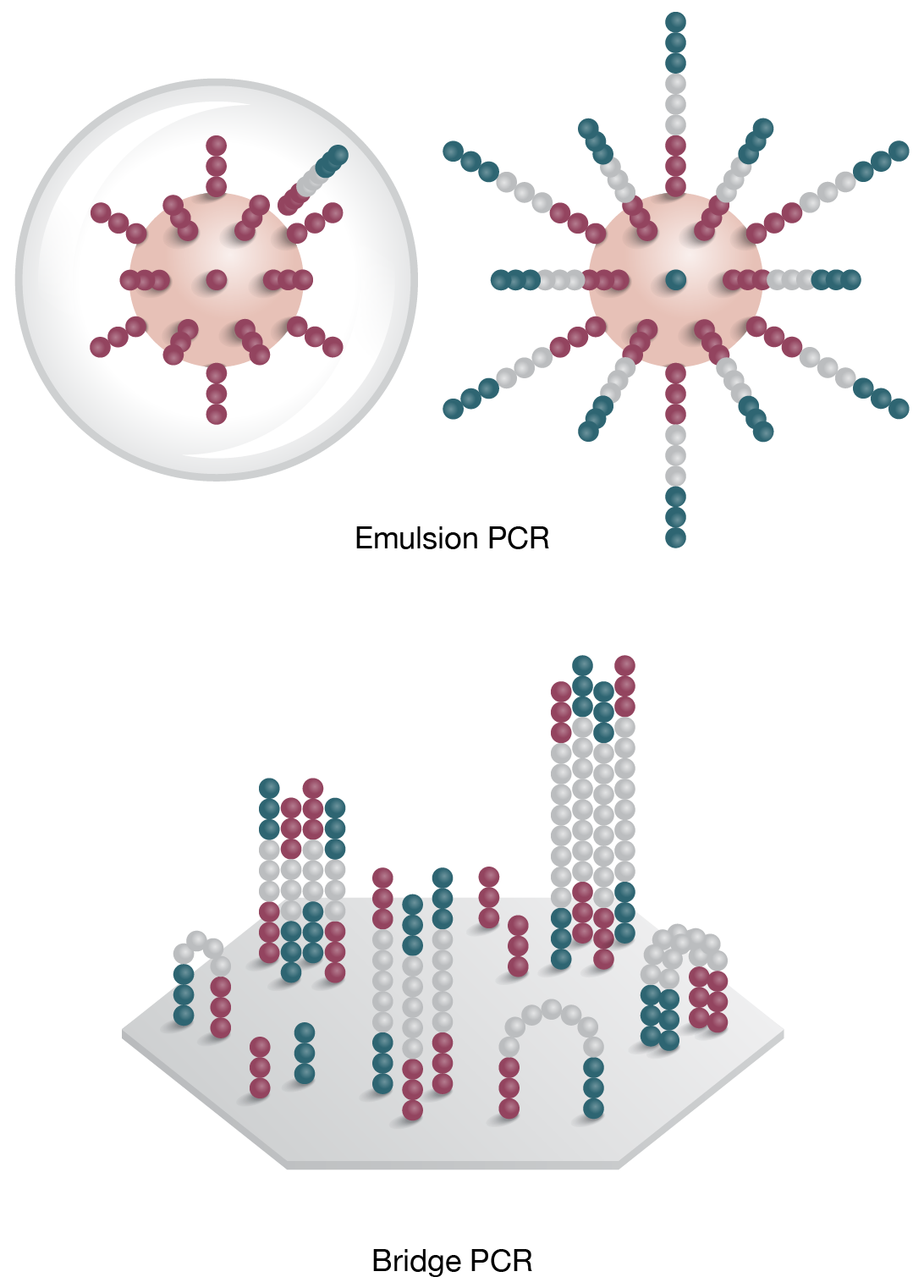
3. Clonal Ampification
Template is PCR amplified by emulsion (on bead) or bridge PCR (solid surface)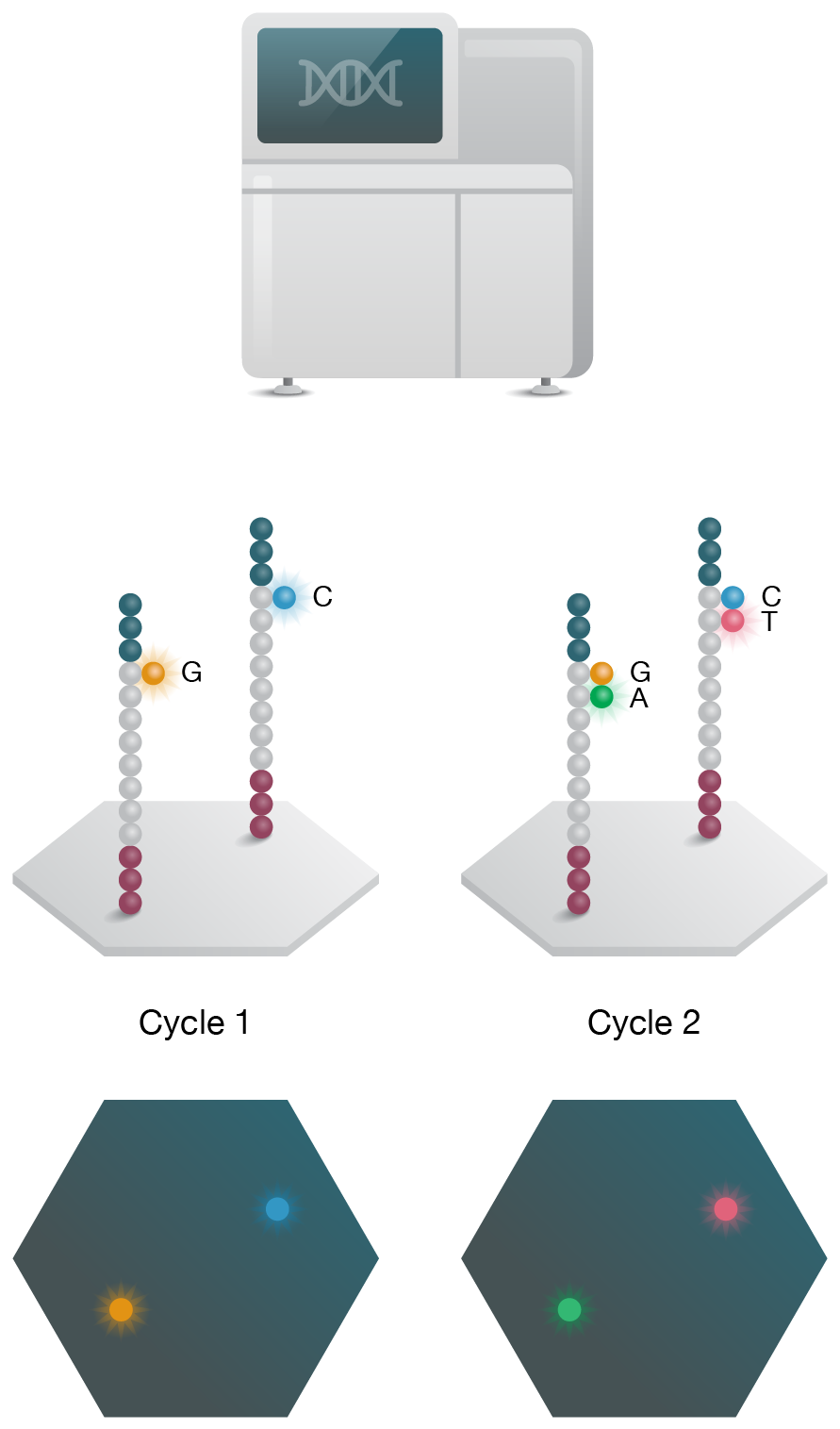
4. Sequencing
Sequencing is performed on NGS instrument and data is analyzed to assess gene function.
Figure 1: Basic NGS RNA-Seq Workflow.
RNA-Seq vs. Microarray Transcriptomics
Over the last decade, RNA-Seq has quickly gained dominance over microarrays due to several key advantages that allow applications beyond differential gene expression. While microarrays require prior knowledge of target sequence to design the hybridization probes, RNA-Seq methods read the cDNA sequence during de novo synthesis; thus, RNA-Seq can be used to study novel transcripts, gene fusions, and non-coding regions. RNA-Seq also offers significantly better dynamic range, which is ideal for detecting low-abundance transcripts. Additionally, non-specific hybridization and other specificity issues are less of a concern in RNA-Seq experiments.
RNA Library Generation
After preparing high-quality RNA for sequencing, the next critical step is library generation. In this step, RNA is converted to cDNA, fitted with platform-specific adaptors, and amplified prior to sequencing. The adaptor sequence is tailored to each sequencing platform and is required for clonal amplification and priming the sequencing reaction. Optional sequence tags or priming sites may also be included depending on the specific application.
There are a few strategies for adding adaptors to cDNA. The most common method uses Y-adapter PCR to add the adapter to blunt-ended cDNA, after the addition of adenosine overhangs. Some kits use RT/PCR to add the adapter sequence to cDNA during the first-strand and second-strand synthesis steps. Other commercial kits use ligation procedures to ligate adaptors to the RNA molecule. The adaptors also serve as priming sites for first- and second-stand cDNA synthesis.
View our NGS Library Preparation products »
RNA Library Strandedness
In a standard protocol, the resulting cDNA library is a mix of two populations: the molecules that originate from the first-strand synthesis and those originating from the second-strand synthesis. As a results strand information as it relates to the original RNA molecule is lost in downstream sequencing. Preserving "strandedness" is useful in studies involving overhanging transcripts, but is not as crucial in differential gene expression analysis. In stranded, or directional, RNA-Seq, a modified cDNA synthesis protocol is used that allows for removal of the second strand prior to PCR amplification.
Our SEQuoia Complete Stranded RNA Library Prep Kit is a high-performance RNA-Seq kit that utilizes a proprietary engineered enzyme to couple cDNA synthesis with adapter addition in a continuous synthesis reaction. This process effectively captures all types and sizes of RNA species in a single library while preserving strandedness, significantly improving the diversity and quality of RNA libraries, even from limited or degraded RNA samples.
Learn more about the SEQuoia Complete Stranded RNA Library Prep Kit »
RNA Library Quantification
Accurate quantification of RNA libraries prior to sequencing on NGS platforms is essential to achieve optimal data and maximize sequencing throughput. Common library quantification methods include UV absorption spectrophotometry, fluorometric spectroscopy with intercalating dyes, real-time quantitative PCR (qPCR) with hydrolysis probes, and more recently, Droplet Digital PCR (ddPCR).
Precise quantification after adapter ligation, before the clonal amplification step, is important for reducing polyclonal reads, which are unusable, and for ensuring proper balancing of multiple libraries pooled together for a single sequencing run. Inadequate balancing of pooled libraries will result in over-representation of some libraries and under-representation of others.
NGS library quantification methods using qPCR are laborious and have significant limitations that make library calibration unreliable (Laurie 2013). NGS library quantification using ddPCR has proven to be highly sensitive and of comparable or better accuracy than previous methods (Robin 2016). The ddPCR library quantification assay generates data plots that are rich with qualitative library information, discerning features such as well-constructed library fragments from adapter-adapter dimers, a capability not available with other NGS library quantification methods.
Learn more about our Digital PCR Library Quantification Kits »
RNA-Seq Technologies
The main NGS technology used in RNA-Seq studies is a short-read sequencing-by-synthesis protocol that allows real-time monitoring of chain extension as a complementary DNA strand is polymerized along the template DNA (cDNA in an RNA-Seq experiment). In this technique, the four fluorescently labeled dNTPs are introduced in turn and imaged upon incorporation into the elongating strand. The number of counts from each cDNA template is then associated with gene abundance during data analysis. As next-generation sequencing technologies have matured over the last decade, a number of variations in RNA-Seq methods and instrumentation has followed, including new long-read and direct RNA-Seq technologies (Fig 2), each with unique advantages for new research applications (Stark 2019).
A. Sequencing by synthesis (Short Read)

B. Single-molecule real-time (Long Read)
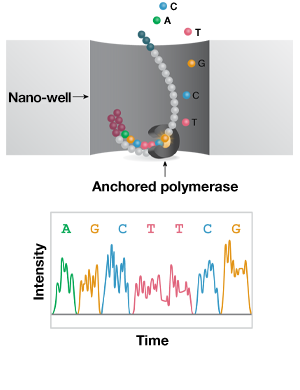
C. Nanopore (Direct Long-Read)

Figure 2: Short- and long-read NGS platforms. A) In the industry-leading short-read NGS platform, template DNA is amplified by bridge PCR to generate clusters on a flow cell. In each reaction cycle, fluorescent nucleotides are introduced, generating a color signal when incorporated into the growing strand. B) In single-molecule real-time (SMRT) long-read sequencing, individual molecules of adapter-ligated template are bound to a single polymerase molecule that is fixed to the bottom of a nanowell. During DNA polymerization of the complementary strand, the wells are imaged to identify the incorporated fluorescent nucleotide. C) In nanopore single-molecule long-read sequencing, the template is ligated to a motor protein and delivered to a flow cell. Here, the motor protein threads the template through a nanopore, where each nucleotide is identified by a current change.
Short-Read RNA-Seq
Over 90% of publicly available RNA-Seq data is generated using high-throughput sequencing-by-synthesis short-read technologies that read 50–500 bp fragments to an average depth of 20–30 million reads per sample. This protocol involves mRNA extraction and cDNA generation, followed by adapter ligation and PCR amplification to generate clusters on a flow cell. During the sequencing reaction, fluorescently labelled nucleotides are introduced in turn, and the growing complementary strand is imaged following each cycle. Computational methods are then used to identify individual transcripts and measure abundance in differential gene expression studies. While short-read RNA-Seq offers very high throughputs and an extensive range of protocols (including those for degraded RNA), there is inherent risk of bias due to the cDNA synthesis and PCR amplification steps. Additionally, this method is not well suited to analyzing isoforms and long transcripts.
Long-Read RNA-Seq
Long-read (1–50 kb) technologies allow single-molecule sequencing of full-length transcripts and are well suited to more complex analyses, like novel isoform discovery. The leading long-read technology is a single-molecule real-time (SMRT) platform. Here, RNA is converted to cDNA using a template-switching reverse transcriptase and PCR amplified to generate sufficient template. The sequencing reaction takes place on a chip, where individual cDNA molecules bind a single polymerase molecule that is fixed to the bottom of a nanowell. The well is imaged following incorporation of each fluorescently labelled nucleotides to the growing strand and the sequence is assembled and interpreted computationally.
Despite the advantage of detecting isoforms without the need to assemble transcripts from short reads, this technology is significantly lower in throughput (500,000‒10 million reads per run), which is limiting for large-scale differential expression analysis studies. It also carries the risk of introducing bias and errors in the reverse transcription and PCR amplification steps – the associated error rates are two orders of magnitude higher than the best short-read methods. Additionally, unlike short-read methods, this protocol is not amenable to studies with degraded RNA.
Direct-Read RNA-Seq
One of the inherent caveats to short- and long-read protocols is the bias introduced by the cDNA synthesis and PCR amplification steps. Nanopore sequencing is an alternate long-read NGS technology that circumvents these workflow steps by directly sequencing the template RNA. In this method, individual template RNA molecules are delivered to a flow cell after library preparation, and docked to a nanopore. Here, the action of a motor protein feeds the RNA strand through the pore, where different nucleotides are detected by a specific change in the current that exists in the pore. Nanopore direct-read RNA-Seq shares many of the advantages and disadvantages of regular long-read methods. Aside from reduced bias, one unique benefit of direct-read technology is that it detects epigenetic modifications to RNA such as methylation.
Single-Cell RNA-Seq
Traditional sequencing protocols performed on templates extracted from cell mixtures do not capture the genomic variation of individual cells. Single-cell sequencing of DNA or RNA captures these differences and is used to investigate new questions in biology. Single-cell methods first require the isolation of single cells by various means including physical manipulation such as micropipetting in tubes, microfluidic methods such as flow sorting or droplet capture, applied electric fields, and optical techniques such as optical tweezers, with distribution into nanowells and in situ barcoding of cells (Shinde 2018). Following library generation, the template is sequenced on an NGS platform.
Single-cell RNA-Seq (scRNA-Seq) is an emerging technology that captures the unique gene-expression signatures of individual cells, as opposed to bulk RNA-Seq methods that report average abundances. In transcriptomics, scRNA-Seq has been used to study gene interaction networks, lineage tracing, and rare cell populations, with applications in immunology and tumor heterogeneity (Hwang 2018; Stark 2019). Since its initial publication in 2009, significant advances in microfluidics and in situ barcoding have evolved the field from studies limited to a handful of cells at a time to large-scale scRNA-Seq studies of organs and entire organisms; two examples of such undertakings are the Human Cell Atlas and the NIH Brain Initiative.
Learn more about our Single-Cell Sequencing products »
Single-Cell ATAC-Seq
The assay for transposase-accessible chromatin by sequencing (ATAC-Seq) is a recently developed technique used to investigate relationships between gene expression and the local state of chromatin accessibility across the entire genome. Actively transcribed DNA is found in open chromatin regions. ATAC-Seq distinguishes these open regions by tagging them using a transposase to insert sequencing adapters into accessible DNA, which is then isolated and used to prepare NGS libraries.
The number of sequencing reads for each region is then used to identify regions of increased accessibility; more reads from a particular region correlates with more open chromatin there. Additional analysis can be used to identify transcription factor binding sites and nucleosome positions (Buenrostro 2015).
Single-cell ATAC-Seq assays provide a novel method for mapping genome-wide chromatin accessibility for thousands of individual single cells, enabling investigation of cell-to-cell epigenetic variations. Our ddSEQ Single-Cell ATAC-Seq Library Prep Kit, for use with the ddSEQ Single-Cell Isolator, provides a simple one-day workflow with high cell throughput, enabling easy access to epigenetic information, including transcriptional dynamics from cell to cell.
Applications of RNA-Seq
RNA-Seq has revolutionized transcriptomics research, allowing scientists to ask new questions about functional genomics as it relates to normal and disease biology. In addition to differential gene expression, the applications of RNA-Seq have quickly expanded to complex RNA biology, clinical diagnostics, nutrition, plant science, and more (Byron 2016). A few examples are provided below.
Alternative Splicing
RNA splicing involves over 90% of coding genes and leads to proteome diversity. Similarly, mis-splicing events can lead to gene malfunction, enabling disease pathways. RNA-Seq technologies have addressed many of the limitations of previous methods (ESTs, RT-PCR, and microarray) for analyzing alternative splicing, with respect to throughput, background noise, and ability to identify novel splicing events. Advancements in RNA-Seq protocols and computational tools have led to genome-wide studies of alternative splicing in mammals, and opportunities in disease and drug development (Zhao 2019). In a 2019 study, Zhou et al. investigated the role of a serine/arginine-rich splicing factor (SRSF) in tumor progression, and identified associated splicing events using RNA-Seq (Zhou 2019).
Rare Gene Variants
Studies of genetic variation within populations have important implications in health and disease, especially diseases without a known cause. Mohammadi et al. used allelic tissue- and population-level RNA-Seq datasets to develop a statistical method for identifying rare variants (outliers). Applying this method to datasets from 70 patients with rare Mendelian muscular disease, they verified the dosage imbalance reported in previous tests, and made some new diagnoses (Mohammadi 2019).
Differential Gene Expression
RNA sequencing is an indispensable tool for understanding human diseases like cancer and infectious disease, with many emerging applications as a clinical diagnostic. It is routinely used to identify gene fusions and differential expression of clinically significant transcripts, yielding significantly better dynamic range compared to microarrays and RT-qPCR methods. Additionally, unlike microarrays and qRT-PCR methods, RNA-Seq can detect and quantify rare or novel transcripts. A comparison study of RNA-Seq and microarray expression analysis demonstrated the reliability of RNA-Seq for evaluating the expression signatures of clinically relevant breast cancer genes (Fumagalli 2014).
Somatic Variants
RNA-Seq can also be used to detect somatic variants such as gene fusions, some of which have a clinical role in disease. For example, several variants in the BCR–ABL1 gene fusion are associated with poor treatment outcomes in patients with chronic myeloid leukemia. In a recent study, Cavelier et al. successfully used long-read RNA-Seq to quantify the clonal distribution of BCR–ABL1 mutations, even in low-abundance samples, in addition to detecting mutations and splice isoforms that were missed by standard clinical protocols (Cavelier 2015).
Infectious Disease
Although RT-qPCR assays are routinely used in the clinic to detect and identify RNA viruses (e.g., HIV, Ebola, and West Nile), RNA-Seq methods are showing utility as a clinical tool for infectious disease. During the 2014 Ebola outbreak in West Africa, RNA-Seq was used to identify genetic variation in 99 viral genomes across 78 patients and elucidated the origin and transmission patterns of the virus (Gire 2014).
Noncoding RNA
The majority of RNA-Seq studies focus on mRNA; however the methodology is also applicable to noncoding RNA (e.g., miRNA, lincRNA, and circRNA), which have diverse regulatory roles in both normal and disease biology. Micro RNAs (miRNAs) have been extensively studied, with an increasing rate of sequence deposition to the public database miRBase, thanks in part to RNA-Seq. In a recent RNA-Seq study, Zhou et al. identified a novel microRNA in a decoction prepared from the honeysuckle plant that directly targeted the influenza A virus, revealing a potential active agent for this ancient Chinese medicine (Zhou 2015). RNA-Seq protocols are also being adapted for other types of noncoding RNA.
Single-Cell Expression Profiling
Beyond bulk analyses of the transcriptome, there are a plethora of questions that can only be explored using single-cell methods (Hwang 2018). Although scRNA-Seq is not as mature as other methods with respect to protocols and data analysis, it is a fast-evolving field that has opened the door to studies of gene- interaction networks, cell lineage pathways, and rare or complex cell populations, like immune cells. An scRNA-Seq study of mouse bone-marrow-derived dendritic cells revealed extensive heterogeneity in mRNA abundance and splicing (Shalek 2013). scRNA-Seq has also been an invaluable tool for dissecting the complex biology of the brain with respect to development. Potential applications also exist in cancer research, where scRNA-Seq is used to study tumor heterogeneity and the mechanisms that lead to metastasis and drug tolerance.
Conclusion
Rapid development in protocols and data analysis over the last decade have made RNA-Seq an invaluable tool for understanding gene function and dysfunction, and made possible large-scale transcriptome surveys by international consortia like the Encyclopedia of DNA Elements (ENCODE) and The Cancer Genome Atlas (TCGA). Future efforts will see protocol improvements to long-read and single-cell RNA-Seq, in addition to data processing and analysis, making these methods more routine and cost-effective for labs.
Related Topics and Products

Next-Generation Sequencing
Learn about next-generation sequencing (NGS) methods, the NGS workflow, and key technologies in the development of large-scale genomic sequencing.
RNA-Seq Workflow
A guide to the steps of an RNA-Seq workflow including library prep and quantitation and software tools for RNA-Seq data analysis.
The Non-Coding Transcriptome
Explore the diverse classes of noncoding RNA, the origins of ncRNA species, their biological roles, and clinical implications.

PCR (Polymerase Chain Reaction)
Learn how PCR works, how to choose the right PCR instrument, how to design, optimize, analyze, and troubleshoot PCR assays.

Real-Time PCR (qPCR)
Discover an array of solutions to optimize, execute, and troubleshoot MIQE-compliant real-time PCR experiments.

Introduction to Digital PCR
Digital PCR is a breakthrough technology that provides ultrasensitive and absolute nucleic acid quantification.
Next-Generation Sequencing
Novel products for NGS library preparation and quantitation used in cutting-edge applications such as whole-transcriptome RNA-Seq and single-cell sequencing.

lncRNA RT-qPCR Workflow
This innovative workflow enables highly sensitive detection of long noncoding RNA (lncRNA) and helps to overcome challenges associated with IncRNA expression analysis.

Traditional PCR Systems
DNA amplification instruments range from personal thermal cyclers to the flexible 1000-series.

qPCR Detection Systems
These systems deliver sensitive, reliable detection of both singleplex and multiplex real-time PCR reactions.

PCR & qPCR Reagents
A wide range of reagents for reverse transcription, PCR, and real-time PCR, optimized to generate accurate and reproducible data.

PCR Plastic Consumables
A large selection of thin-wall PCR tubes, plates, seals, and accessories precisely manufactured for optimal fit and cycling performance in Bio-Rad thermal cyclers, real-time PCR systems, ddPCR systems, and all major competitor systems.

PrimePCR PCR Primers, Assays & Arrays
Experimentally validated PCR primer and probe assays for gene expression, copy number variation, and mutation detection analysis for real-time PCR and Droplet Digital PCR.
Droplet Digital PCR Assays
Bio-Rad offers a comprehensive portfolio of Digital PCR Assays and Kits for numerous applications including Mutation Detection, Copy Number Determination, Genome Edit Detection, Gene Expression, Expert Design Assays, Residual DNA Quantification and Library Quantification.
References
Bik EM. The Hoops, Hopes, and Hypes of Human Microbiome Research. Yale J Biol Med. 2016 Sep; 89(3):363-373. PMCID: PMC5045145.
Buenrostro JD et al. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Current Protocols in Molecular Biology. 2015;109: 21.29.1–21.29.9. DOI: 10.1002/0471142727.mb2129s109. PMCID: 4374986.
Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet. 2016 May;17(5):257-71. DOI: 10.1038/nrg.2016.10. PMID: 26996076.
Cavelier L, Ameur A, Häggqvist S, et al. Clonal distribution of BCR-ABL1 mutations and splice isoforms by single-molecule long-read RNA sequencing. BMC Cancer. 2015;15:45. DOI: 10.1186/s12885-015-1046-y. PMCID: PMC4335374.
Cheng M, Cao L, Ning K. Microbiome Big-Data Mining and Applications Using Single-Cell Technologies and Metagenomics Approaches Toward Precision Medicine. Front Genet. 2019 Oct; 10:972. PMCID: PMC6794611.
Encyclopedia of DNA Elements (ENCODE): https://www.encodeproject.org/.
Fumagalli D, Blanchet-Cohen A, Brown D, et al. Transfer of clinically relevant gene expression signatures in breast cancer: from Affymetrix microarray to Illumina RNA-Sequencing technology. BMC Genomics. 2014;15:1008. DOI: 10.1186/1471-2164-15-1008. PMCID: PMC4289354.
Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345(6202):1369-72. DOI: 10.1126/science.1259657. PMCID: PMC4431643.
Human Cell Atlas: https://www.humancellatlas.org/.
Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018 ;50(8):96. DOI: 10.1038/s12276-018-0071-8. PMCID: PMC6082860.
Laurie MT et al. Simultaneous digital quantification and fluorescence-based size characterization of massively parallel sequencing libraries. Biotechniques 2013;55, 61–67. DOI: 10.2144/000114063. PMCID: PMC4018218.
Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T. Transcriptomics technologies. PLoS Comput Biol. 2017;13(5):e1005457. DOI: 10.1371/journal.pcbi.1005457. PMCID: PMC5436640.
Mohammadi P, Castel SE, Cummings BB, et al. Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science. 2019;366(6463):351-356. DOI: 10.1126/science.aay0256. PMCID: PMC6814274.
NIH Brain Initiative: https://braininitiative.nih.gov/.
Robin JD, Ludlow AT, LaRanger R, Wright WE, Shay JW. Comparison of DNA Quantification Methods for Next-Generation Sequencing. Sci Rep. 2016 Apr 6;6:24067. DOI: 10.1038/srep24067. PMCID: PMC4822169.
Shalek AK, Satija R, Adiconis X, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498(7453):236-40. DOI: 10.1038/nature12172. PMCID: PMC3683364.
Shinde P, Mohan L, Kumar A, et al. Current Trends of Microfluidic Single-Cell Technologies. Int J Mol Sci. 2018; 19(10):3143. DOI: 10.3390/ijms19103143.
Soneson C et al. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat Commun 10, 3359 (2019). DOI: 10.1038/s41467-019-11272-z.
Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20(11):631-656. DOI: 10.1038/s41576-019-0150-2. PMID: 31341269.
The Cancer Genome Atlas (TCGA): https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
Wang Y et al. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol 39, 1348–1365 (2021). DOI: 10.1038/s41587-021-01108-x
Zhao S. Alternative splicing, RNA-Seq and drug discovery. Drug Discov Today. 2019 Jun;24(6):1258-1267. DOI: 10.1016/j.drudis.2019.03.030. PMID: 30953866.
Zhou Z, Li X, Liu J, et al. Honeysuckle-encoded atypical microRNA2911 directly targets influenza A viruses. Cell Res. 2015;25(1):39-49. DOI: 10.1038/cr.2014.130. PMCID: PMC4650580.
Zhou X, Wang R, Li X, et al. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. J Clin Invest. 2019;129(2):676-693. DOI: 10.1172/JCI120279. PMCID: PMC6355305.