
In 1972, Walter Fiers and his team were the first to determine the complete DNA sequence of a gene, the coat protein of bacteriophage MS2 (Min Jou 1972). Since then, the toolbox of experimental techniques that can be applied to understanding how DNA, RNA, and proteins function has been vastly expanded. In the past two decades, the speed of DNA sequencing has increased exponentially, and the cost has fallen similarly, enabling the sequencing of entire genomes. Parallel advances in computing led to methods for the analysis of these unprecedented amounts of biological data and the birth of the field of bioinformatics. Initial efforts in sequence analysis focused on the identification of protein coding sequences, with the surprising result that these typically comprise less than 2% of a whole genome.
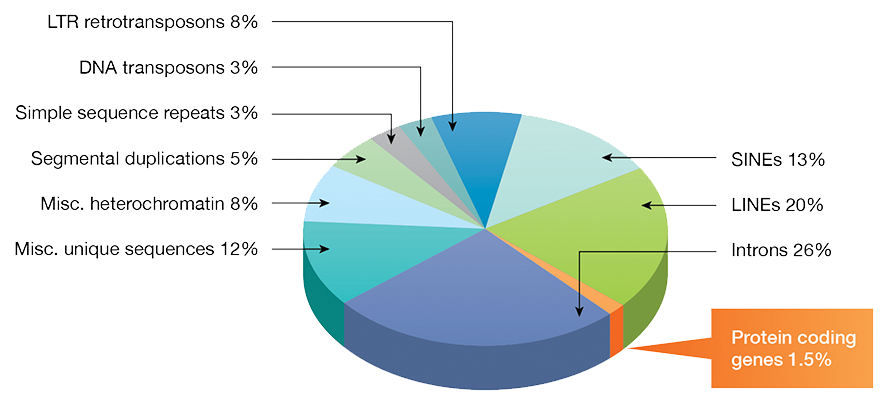
Fig. 1. Fractions of DNA sequence types in the human genome.
Upon identifying a sequence of interest, the following step in the discovery process is to understand its function. Whether the expression of this sequence leads to a protein or to a non-coding transcript, the function of the final product needs to be understood to validate any hypothesis around the role it plays in any given phenotype. Here, we discuss the tools for investigating the function of genes in vitro, in silico, and in model organisms.
Functional Prediction Based on Theoretical Protein Structure
A protein coding sequence represents the chain of amino acids that make up a given protein, known as primary protein structure. The function of the protein is determined by how the amino acid chain folds into 3D structures. Elucidating protein structure can be a difficult process, traditionally requiring protein crystallization followed by x-ray crystallography. Instead of using physical methods to resolve protein structure, bioinformatics techniques can be used to determine a theoretical structure, which can then be used to predict function. Bioinformatics resource pages, such as ExPASy and EMBL-EBI have many freely available programs that can be used for sequence and structural analysis.
Homology-Based Predictions
Typically, functional analysis based on protein sequence starts with a search for other proteins with similar sequences. Proteins with homologous function often also share similar sequences and reflect common ancestry. Similar protein sequence would suggest a similar structure, and therefore result in similar function. Protein sequence searches can also uncover orthologs, which are genes in different species that evolved from a common ancestral gene. For example, the human Histone H1 protein shares high sequence similarity to the mammalian Histone H1 orthologs and also have conserved function as a DNA packaging protein. Protein sequence searches and alignments are able to be performed through the Basic Local Alignment Search Tool (BLAST), available for public use through the National Center for Biotechnology Information (NCBI).

Fig. 2. Protein sequence alignment showing common sequence motifis.
Protein Domain- and Motif-Based Predictions
Protein 3D structure is complex and multileveled. Many proteins contain common sequence patterns that give rise to secondary structures such as alpha helices or beta sheets. These secondary structures may then be arranged into larger super-secondary structures called motifs, which in turn can be combined to form tertiary structures known as domains. Many proteins consist of multiple structural domains linked together like building blocks to create one functional unit, for example, a transcription factor containing a DNA-binding domain, a signal-sensing domain, and an activator domain.
Comparing primary sequence similarity to known proteins may not always reveal protein function, but sequence searches may find subregions encoding structural domains, motifs, or signaling sequences that can inform functional roles. For example, a nuclear localization signal suggests that a protein has a role in the nucleus. Protein domain searches can be performed on websites such as PROSITE, which consists of a database of protein domains, families, and functional sites.
The identification of domains allows for a more specific homologous sequence search using just the amino acid sequence of the domain as the query. This can reveal homologies with proteins that were not found when querying the entire amino acid sequence and help to determine a more specific function.

Fig. 3. Protein domains shared by proteins with varying functions.
Comparative Protein Modeling
When the protein of interest has sequence similarity to a protein with a known structure, an in silico amino acid mapping can be performed to generate a theoretical protein structure using techniques called homology modeling and protein threading. Homology modeling relies on the assumption that homologous proteins will share a similar structure due to the fact that protein folds are more evolutionarily conserved than sequence. Because of this, a sequence of interest can be modeled even on a distantly related homolog's structure. Protein threading is very similar to homology modeling in that an amino acid sequence is mapped onto the structure of another protein, or protein domain. However, in protein threading, the sequence is scanned across a database of known protein structures in an effort to find a compatible structure. This method is highly reliant on a solid sequence alignment program and a scoring system to measure “fitness”. When a probable 3D structure is established, function may be inferred from amino acids found in binding pockets, functional folds, new found domains, or similarity to other proteins.
De Novo Protein Structure Prediction
When structures are not found through sequence homology or comparative modeling, computational biology can be used to predict tertiary structure. De novo protein structure prediction uses algorithms and deep learning to predict structure based solely on amino acid sequence. This method tends to require a vast amount of computational power because the predictors function by producing candidate conformations and then choosing amongst them based on their thermodynamic stability and energy state. Because of the computing power needed, only small protein structures are typically predicted in this way.
Genomics Studies In Vitro versus In Vivo
Gene function is relative, based on the context of the cell in which it is expressed. Function will vary in different cell types due to differences in the cellular microenvironment. Choosing the right model system for functional genomics studies is critical to reveal the true effects of a gene or genomic sequence. In vitro studies provide a fast and cost-effective approach for the study of genes on a cellular level. In contrast, in vivo studies add great value by enabling the observation of gene function in an entire organism, although these studies can be much more time-consuming and costly.
Model Organisms for In Vivo Experimentation
All of the model organisms below have had their entire genomes sequenced, allowing functional genomics to dive deeper and achieve more comprehensive analyses.
- Arabidopsis – Related to cabbage and mustard, Arabidopsis thaliana is the model organism of choice for many plant biologists. The genome is relatively small and easily manipulated. They can be cross-breed but are also self-fertilize and can produce thousands of seeds, with a generation time of only six weeks. While Arabidopsis is not an important agricultural plant, it is related to crop plants, and its research has contributed to more sustainable and high-yield agriculture.
- Yeast – Yeast is one of the simplest eukaryotic single-celled organisms. With 6,000 genes and a doubling time of ~100 minutes, Saccharomyces cerevisiae, also known as baker’s yeast, is widely used as a model eukaryote with the ease of culture of a microbe. Due to its fast replication time and relatively easy genetic manipulation, yeasts are often used to study basic cellular processes like DNA repair, telomere maintenance, and cell division.
- C. elegans – The nematode worm, Caenorhabditis elegans, has a total of approximately 1,000 somatic cells (nearly one-third are neurons) and are completely transparent, making them easy to study under a microscope. C. elegans live for about two weeks, have many human gene homologs, and are easily genetically altered. For these reasons, they are often used to study gene function on longevity and the nervous system.
- Zebrafish - The zebrafish, Danio rerio, is a vertebrate that shares 70% of its genes and the same major organs with humans. Zebrafish can produce hundreds of offspring per week, which are transparent and develop outside the body, making them great model organisms for studying embryonic development. They have also been used to study cardiovascular development and disease, as their hearts and vasculature are similar to humans.
- Drosophila - The common fruit fly, Drosophila melanogaster, has only four chromosomes yet it shares 60% of its genes with humans. They reproduce quickly, with a life cycle of about 12 days. Males and females are easily separated, which enables easy breeding to develop novel genetic cross-breeds. Drosophila are used to study a wide range of biological processes including developmental differentiation and sexual reproduction.
- Mice - Mice, Mus musculus, are mammals with close genetic and physiological similarities to humans. They are the premiere tools for studying complex physiological systems that mammals share, such as the immune, endocrine, and nervous systems. Mice also naturally acquire similar diseases as humans, such as cancer, diabetes, and addiction. Manipulation of the mouse genome is a fairly straightforward process, made easier by recent advances such as CRISPR-Cas genome editing. Many genetically modified mouse lines are readily available for novel research, created over decades of functional genomic research in mice.
Options for In Vitro Experimentation
- Cell Lines – Thousands of cell lines from all types of model organisms and humans have been created or isolated and are readily available for novel research. Many human cell lines are derived from cancer cells which allows them to divide continuously. Cells can also be immortalized through the addition of genes that inhibit cellular senescence and activate cell division (often through addition of viral genes). While cell lines are an inexpensive and easy way to conduct genetic research at the cellular level, it is imperative to be aware that they are not normally functioning cells. It is often recommended to confirm functional genomics results among several disparate cell lines to ensure that the result is not just a cell line effect.
- Primary Cells Primary cells are harvested directly from a whole organism (mostly mouse or human) and do not divide continuously. They are sometimes very difficult to culture and can be expensive but are much more representative of real tissue responses to genetic disturbances. Great care needs to be taken when working with primary cells, as they can be very sensitive to their environment or certain reagents. It is recommended to seek out established protocols for genetic research in primary cells.
- Human Stem Cells Stem cells can be used to study the function of genes in many different cell states due to their pluripotency, the ability to differentiate into various cell types. Gene function during embryonic development, tissue differentiation, and in maintaining pluripotency can all be studied using stem cells.
- Organoids Organoids are 3D self-organizing structures derived from stem cells that mimic the features of whole organs such as a liver or kidney. With recent advances in CRISPR-Cas gene editing, organoids can be generated that harbor specific genomic mutations. These organoids can then be used to study organ development or be used as models for disease.

Fig. 4. Examples of organoids cultured from tissue-specific cells.
Tissue Expression Dynamics
Genes are often only expressed in some tissues and are silenced in the rest of the body. However, normally silenced genes can be awakened under certain conditions, such as stress, infection, or disease. To achieve a comprehensive analysis of gene function, a range of tissues need to be examined. Gene expression is often determined by mRNA quantification; however, mRNA is not always translated, and protein stability can play a role in function. It is therefore recommended to examine both mRNA and protein levels in a panel of tissues for functional studies.
Reverse Genetics
Reverse genetics is a method used to help understand the function of a gene by analyzing the phenotypic effects of specific engineered gene sequences. Reverse genetics usually proceeds in the opposite direction of so-called forward genetic screens of classical genetics. In other words, while forward genetics seeks to find the genetic basis of a phenotype or trait, reverse genetics seeks to find what phenotypes arise as a result of particular genetic sequences. Experimentally, this question can be addressed by adding or removing the sequence, by upregulating it, or inactivating it partially or fully, then observing differences in the functional phenotypes of the transformed cells or organism. The genome can be directly altered, or RNA transcripts can be silenced to block the expression of a specific genomic sequence. Typically, reverse genetics is used to uncover the function of proteins; however, non-coding RNA has also been shown to have important cellular functions and can be studied using the same techniques. Gene knockout, knock-in, overexpression/upregulation, and knock-down are all techniques used in reverse genetics to silence or change the sequence of specific genes.
Early studies in reverse genetics used a variety of techniques to alter gene sequences, including random mutagenesis followed by intensive screening, the use of viral vectors and Agrobacterium for transformation, directed transposon mutagenesis, and other naturally occurring mutagenic phenomena like repeat-induced point mutation. Later, a gene-targeting method was developed to introduce defined genetic alterations into the host genome. This method exploits homologous recombination, an innate cellular DNA repair mechanism, to integrate an engineered DNA construct bearing the modified gene sequence flanked by DNA with sequence homologous to the target genetic locus on the host chromosome. This gene-targeting method is much more selective than random mutation; however, screening of the transformed cells is still necessary due to multicopy integration and off-target (ectopic) integration of the constructs.

Fig. 5. Gene editing. A short guide RNA can be used to target Cas9 to a sequence of interest. Cas9 introduces a double strand DNA break to allow highly efficient gene editing.
More recently developed genetic engineering tools offer more precise methods of integrating exogenous DNA into the host chromosome at desired targets: the use of zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the CRISPR-Cas system. All these methods direct dsDNA cleavage of the genome at specific sequences, and can then introduce random mutations or guide insertion or deletion of a target sequence. Both ZFNs and TALENs are proteins that cut DNA at specific sequences guided by distinct DNA-binding motifs. In contrast, the CRISPR-Cas system is able to cut specific DNA sequences using direction from guide RNA (gRNA). Collectively, these modern techniques are known as targeted genome editing.
Learn more about Targeted Genome Editing »
Gene knockout refers to the physical deletion of a gene from the genome. In the past, homologous recombination was the most widely used method to achieve a targeted gene knockout, but it was difficult and inefficient. Today’s targeted genome editing techniques make knocking out genes relatively easy. The CRISPR-Cas system has become the leading method for knocking out genes due to its high efficiency and low cost.
- Pros: Knocking out a gene ensures that the gene of interest will absolutely not be expressed. In addition, functional validation can be performed by introducing an expression plasmid with the knocked-out gene. Functional restoration of the knocked-out gene of interest by transient expression can confirm functional results observed when the gene was absent.
If knocked out at an embryonic stage, whole organisms can be generated that lack a particular genomic sequence for whole-organism studies. Conditional knockouts can also be created, in which a gene is deleted only in specific cell types. With the efficiency of the CRISPR-Cas system for gene knockout, high-throughput genetic screening using pooled libraries is also possible. - Cons: Non-specific editing of the genome is an issue for all types of knock-out techniques, including CRISPR-Cas. Specificity of DNA cleavage is dictated by sequence homology of the gRNA to the desired genomic sequence. Even strong sequence homology can lead to off-target binding and undesired cleavage. Full genome sequencing may be required to reveal off-target mutations.
Gene knockdown refers to the reduction of gene expression through the degradation of mRNA transcripts. Using the natural RNA interference (RNAi) process, complementary small interfering RNA (siRNA) can be designed to bind to specific mRNA transcripts and recruit RNA degradation complexes. siRNAs are 20–25 bp in length and can be transiently transfected into cells for immediate experimentation. Alternatively, short-hairpin RNA (shRNA), which is processed by the cell into siRNA, can be constitutively expressed in cells through plasmid transfection or by genomic integration via lentivirus.

Fig. 6. Basic molecular mechanism for siRNA silencing of gene expression.
- Pros: Gene knockdown using RNAi is a well-established method that can achieve high efficiency in targeted gene silencing. It is fairly easy to design RNAi targets for your gene of interest, enabling gene knockdown in many cells simultaneously.
- Cons: Since RNAi relies on sequence complementary to the target mRNA and corresponding siRNA, design of the siRNA is critical, and knockdown can often exhibit off-target effects. In addition, this method only reduces the amount of gene expression, but does not completely eliminate it. Sometimes even a small amount of gene expression is enough to retain the functional phenotype. Due to these reasons, several siRNA designs are recommended to target one gene and are often pooled together to achieve more complete gene silencing.
Gene knock-in is a method that introduces new sequences into the genome. Addition of a gene mutation, additional copies of a gene, or a gene expressing a reporter protein such as green fluorescent protein (GFP) are all considered knock-ins. Mouse models of disease are often made by knocking-in mutant alleles and overexpressing genes. The same techniques used in gene knockout can be used for gene knock-in. In addition, gene knock-in can also be achieved using lentiviral delivery; however, this technique lacks specificity in where the new DNA is inserted. CRISPR-Cas gene editing is now the method of choice for knocking in genes due to its efficiency and, in diploid organisms, its ability to modify both alleles of a gene simultaneously.
- Pros: Gene knock-in enables the introduction of new genes into a genome and studies of the effects of a single mutation or multiple gene copies on a cell or organism. Because changes are made directly to the genome, expression of non-mutant genes are not an issue.
- Cons: Off-target effects may occur if the specificity of sequence insertions is imperfect. Due to direct genome editing, whole-genome sequencing is needed to determine the location of desired and undesired insertions.
Understanding DNA and RNA regulation
Transcription and translation are highly regulated processes that are fundamental in the control of the cellular ecosystem. Protein binding to DNA or RNA can either suppress or enhance transcription and translation. Uncovering which proteins are regulators and where they bind to DNA or RNA is critical to our understanding of genomic regulation. Therefore, there are many techniques that have been developed to experimentally find protein-DNA and protein-RNA interactions.
Transcription factors are effector proteins that bind to DNA, acting as either “activators” or “repressors” by recruiting additional cellular machinery to block or activate transcription. These effectors bind to specific sequences within or adjacent to a gene-coding sequence and so are called cis-effectors, with their binding sites called cis-elements. Transcription factor binding, however, is not the only mechanism that effects transcription. Distal DNA sequences, known as trans-elements, can affect both transcription and transcript processing. Trans-elements may be a gene encoding a protein effector such as a transcription factor or other effectors such as microRNA.
Another factor in the high-level control of gene transcription is the chromatin state around the gene of interest. Chromatin is a condensed form of DNA complexed with proteins, called histones, found in the chromosomes of eukaryotic cells. Post-translational modification of certain histones denote areas of relaxed, or open chromatin (euchromatin) or tightly packed chromatin (heterochromatin).
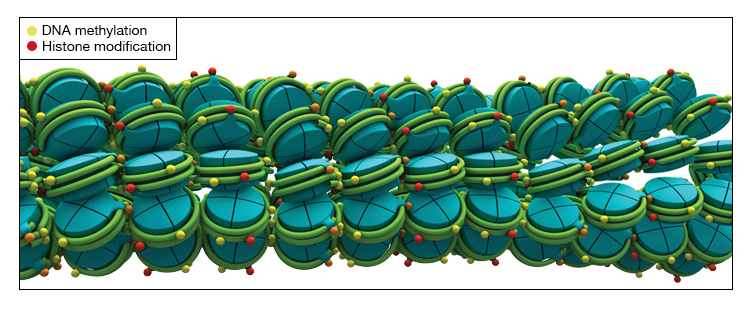

Fig. 7. Structures of heterochromatin and euchromatin DNA-histone complexes.
Euchromatin are areas with active transcription, while heterochromatin is transcriptionally inactive. Questions pertaining to transcriptional regulation of specific genes may be answered by uncovering the transcription factors and modified histones that bind to DNA within and around the gene of interest.
The association between proteins and genomic DNA has an important regulatory role in many cellular processes, including transcription, cell cycle, and epigenetic silencing. The following assays are used to study DNA-protein interactions.
- Chromatin Immunoprecipitation (ChIP)
An assay for identifying the binding site of transcription regulators, histones, and other proteins within the context of the cell - DNA electrophoretic mobility shift assay
An affinity electrophoresis assay that detects binding of proteins to DNA, based on the observed shift of the nucleic acid band - DNA footprinting
Used to identify the location of proteins bound to radiolabeled DNA based on the electrophoretic pattern generated when the complex is treated with DNase - DNA pull-down assay
A DNA probe labeled with an affinity tag is used to pull down target DNA and associated protein(s) from a cell lysate
In both prokaryotes and eukaryotes, gene expression is also regulated by DNA methylation, the addition of methyl groups to DNA without affecting the coding sequence, also known as decoration. In eukaryotes, methylation of cytosine bases in DNA at the 5'-position is linked to a closed chromatin structure. Histone proteins can also be methylated/demethylated or acetylated/deacetylated, triggering changes in chromatin configuration. Collectively, the control of gene regulation by noncoding DNA elements and the noncoding modification of DNA and histones is known as epigenetic control.
Learn more about Epigenetics and Chromatin Structure »
Once mRNA is transcribed from a gene, its translation into protein is also tightly regulated through protein binding. There is an entire class of proteins called RNA-binding proteins (RBPs) responsible for mRNA processing. In eukaryotes, mRNA transcripts are capped, spliced, modified with a Poly(A) tail, and exported from the nucleus for translation in the cytoplasm through RBPs. RBPs can function in alternative RNA splicing or even RNA sequence editing, which results in the expression of various proteins from one gene.

Fig. 8. Variations in RNA splicing patterns can yield different proteins from the same transcript.
Once in the cytoplasm, another layer of mRNA regulation occurs in recruiting or blocking the ribosome to the mRNA for protein translation, along with activation or suppression of mRNA transcript degradation, resulting in a dynamic population turnover of actively translated transcripts. mRNA regulation is very nuanced and has just as significant an effect on gene function as DNA regulation.
Understanding of RNA-protein interactions is vital to how proteins are regulated. The following methods are used to detect RNA-binding proteins.
- RNA immunoprecipitation
A method like chromatin immunoprecipitation (CHIP) for DNA that is used instead to detect the association of proteins with RNA - Fluorescence in situ hybridization
Uses single-stranded fluorescently labeled DNA and RNA probes to detect specific DNA or mRNA sequences in cells and tissue - Exogenous RNA pull-down assay
Selectively pulls down RNA-protein complexes from a sample using in vitro-generated RNA probes carrying a high-affinity tag - Endogenous RNA pull-down assay
Pulls down native RNA-protein complexes from a sample using small RNA probes with a high-affinity tag to target RNA of interest
The Future of Functional Genomics
The human genome project revealed that there are 20,000-25,000 genes in the human genome. This was the first step in understanding humans at a molecular level. Now, the focus is on elucidating the function of each gene on an individual basis, and as a system of interacting molecules. Through the use of modern molecular biology and bioinformatics techniques, functional genomics research is becoming more efficient, allowing for large-scale investigations across whole genomes. CRISPR-Cas gene editing has been a game changer, enabling high-throughput gene knockout screens to reveal the function of many genes simultaneously. The ease and lower cost of sequencing is enabling massive amounts of RNA and DNA data to be analyzed. These data are collected into databases available for “genome mining” that can reveal the genetic basis for disease. Computational literature curation and prediction interactions among proteins, DNA, and RNA are now generating reliable interaction maps with easily accessible information for each gene, transcript, and protein. This information will help to reveal the role of RNA complexes, which have recently been in the spotlight as gene regulators. Overall, the pace of functional genomics is accelerating year over year, giving rise to a detailed understanding of human life and the ability to generate more targeted therapeutics to treat disease.
Related Topics and Products

Next-Generation Sequencing
Learn about next-generation sequencing (NGS) methods, the NGS workflow, and key technologies in the development of large-scale genomic sequencing.
RNA-Seq Workflow
A guide to the steps of an RNA-Seq workflow including library prep and quantitation and software tools for RNA-Seq data analysis.
The Non-Coding Transcriptome
Explore the diverse classes of noncoding RNA, the origins of ncRNA species, their biological roles, and clinical implications.

PCR (Polymerase Chain Reaction)
Learn how PCR works, how to choose the right PCR instrument, how to design, optimize, analyze, and troubleshoot PCR assays.

Real-Time PCR (qPCR)
Discover an array of solutions to optimize, execute, and troubleshoot MIQE-compliant real-time PCR experiments.
Introduction to Digital PCR
Digital PCR is a breakthrough technology that provides ultrasensitive and absolute nucleic acid quantification.
Next-Generation Sequencing
Novel products for NGS library preparation and quantitation used in cutting-edge applications such as whole-transcriptome RNA-Seq and single-cell sequencing.

lncRNA RT-qPCR Workflow
This innovative workflow enables highly sensitive detection of long noncoding RNA (lncRNA) and helps to overcome challenges associated with IncRNA expression analysis.

Traditional PCR Systems
DNA amplification instruments range from personal thermal cyclers to the flexible 1000-series.

qPCR Detection Systems
These systems deliver sensitive, reliable detection of both singleplex and multiplex real-time PCR reactions.

PCR & qPCR Reagents
A wide range of reagents for reverse transcription, PCR, and real-time PCR, optimized to generate accurate and reproducible data.

PCR Plastic Consumables
A large selection of thin-wall PCR tubes, plates, seals, and accessories precisely manufactured for optimal fit and cycling performance in Bio-Rad thermal cyclers, real-time PCR systems, ddPCR systems, and all major competitor systems.

PrimePCR PCR Primers, Assays & Arrays
Experimentally validated PCR primer and probe assays for gene expression, copy number variation, and mutation detection analysis for real-time PCR and Droplet Digital PCR.
Droplet Digital PCR Assays
Bio-Rad offers a comprehensive portfolio of Digital PCR Assays and Kits for numerous applications including Mutation Detection, Copy Number Determination, Genome Edit Detection, Gene Expression, Expert Design Assays, Residual DNA Quantification and Library Quantification.
Citations
Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet. 2017 Nov;18(11):643-658. Epub 2017 Aug 14. DOI: 10.1038/nrg.2017.57. PMID: 28804139.
Aubert P, Suárez-Fariñas M, Mitsui H, Johnson-Huang LM, Harden JL, Pierson KC, Dolan JG, Novitskaya I, Coats I, Estes J, Cowen EW, Plass N, Lee CC, Sun HW, Lowes MA, Goldbach-Mansky R. Homeostatic tissue responses in skin biopsies from NOMID patients with constitutive overproduction of IL-1β. PLoS One. 2012;7(11):e49408. Epub 2012 Nov 30. DOI: 10.1371/journal.pone.0049408. PMCID: PMC3511496.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015 Jul 23;523(7561):486-90. Epub 2015 Jun 17. DOI: 10.1038/nature14590. PMCID: PMC4685948.
Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev. 2018 Sep;174:3-17. DOI: 10.1016/j.mad.2017.12.002. PMID: 29268958.
Cole J, Morris P, Dickman MJ, Dockrell DH. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharmacol Ther. 2016 Nov;167:85-99. Epub 2016 Aug 9. DOI: 10.1016/j.pharmthera.2016.07.013. PMCID: PMC5109899.
Cusanovich, D. A. et al. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). DOI: DOI: 10.1126/science.aab1601.
Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006 Oct;16(10):1299-309. Epub 2006 Sep 5. DOI: 10.1101/gr.5571506. PMCID: PMC1581439.
Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G, Nathan C. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001 Oct 15;194(8):1123-40. DOI: 10.1084/jem.194.8.1123. PMCID: PMC2193509.
Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011 May 19;473(7347):398-402. Epub 2011 Apr 3. DOI: 10.1038/nature10008. PMID: 21460836.
Gavrilov A, Eivazova E, Priozhkova I, Lipinski M, Razin S, Vassetzky Y. Chromosome conformation capture (from 3C to 5C) and its ChIP-based modification. Methods Mol Biol. 2009;567:171-88. Erratum in: Methods Mol Biol. 2009;567:E1. DOI: 10.1007/978-1-60327-414-2_12. PMID: 19588093.
Hamamoto R, Komatsu M, Takasawa K, Asada K, Kaneko S. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules. 2019 Dec 30;10(1):62. DOI: 10.3390/biom10010062. PMCID: PMC7023005.
Hofmann SR, Morbach H, Schwarz T, Rösen-Wolff A, Girschick HJ, Hedrich CM. Attenuated TLR4/MAPK signaling in monocytes from patients with CRMO results in impaired IL-10 expression. Clin Immunol. 2012 Oct;145(1):69-76. Epub 2012 Aug 4. DOI: 10.1016/j.clim.2012.07.012. PMID: 22940633.
Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5(1):1–8. DOI: 10.1016/j.gendis.2018.01.002.
Kita Y, Yonemori K, Osako Y, Baba K, Mori S, Maemura K, Natsugoe S. Noncoding RNA and colorectal cancer: its epigenetic role. J Hum Genet. 2017 Jan;62(1):41-47. Epub 2016 Jun 9. DOI: 10.1038/jhg.2016.66. PMID: 27278790.
Laird PW (2010). Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 3, 191–203. DOI: 10.1038/nrg2732. PMID: 20125086.
Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004 Jan;38(1):32-8. DOI: 10.1002/gene.10250. PMID: 14755802.
Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012 Apr 17;109(16):E962-71. Epub 2012 Feb 27. DOI: 10.1073/pnas.1121568109. PMCID: PMC3341013.
Ohlsson R, Göndör A. The 4C technique: the 'Rosetta stone' for genome biology in 3D? Curr Opin Cell Biol. 2007 Jun;19(3):321-5. Epub 2007 Apr 26. DOI: 10.1016/j.ceb.2007.04.008. PMID: 17466501.
Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010 Oct;11(10):936-44. Epub 2010 Aug 22. DOI: 10.1038/ni.1920. PMID: 20729857.
Shema E, Bernstein BE, Buenrostro JD. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat Genet. 2019 Jan;51(1):19-25. Epub 2018 Dec 17. DOI: 10.1038/s41588-018-0290-x. PMID: 30559489.
Surace AEA, Hedrich CM. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front Immunol. 2019 Jul 4;10:1525. DOI: 10.3389/fimmu.2019.01525. PMCID: PMC6620790.
Torres-Garcia S, Yaseen I, Shukla M, Audergon PNCB, White SA, Pidoux AL, Allshire RC. Epigenetic gene silencing by heterochromatin primes fungal resistance. Nature. 2020 Sep;585(7825):453-458. Epub 2020 Sep 9. DOI: 10.1038/s41586-020-2706-x. PMCID: PMC7116710.