
On This Page |
Chromatin Organization | Methods Used to Study Epigenetics | Clinical Implications of Epigenetics | Related Topics and Products |
Epigenetics is the study of the configuration and chemistry of DNA in chromosomes and changes in gene expression patterns that cannot be traced to the DNA sequence. Unlike phenotypes that are associated with mutations in the underlying DNA code, epigenetic changes involve heritable covalent modifications to chromatin structure, such as DNA methylation and histone modification. Here we highlight common epigenetic markers, the techniques used to study them, and their role in biology and human disease. Non-coding RNAs (e.g., microRNA), also involved in epigenetic regulation, are discussed in detail in The Coding and Non-Coding RNA.
Chromatin Organization
DNA is stored in a highly structured complex called chromatin. Epigenetic processes control gene expression by altering chromatin structure (Figure 1). Actively transcribed genes are associated with accessible chromatin regions, while transcriptionally silent genes are found in inaccessible chromatin regions. These modifications made to DNA and proteins, which impact chromatin structure, are referred to as epigenetic markers (or marks) and subsequently inherited as they are passed on through rounds of cell division.
- A

- B
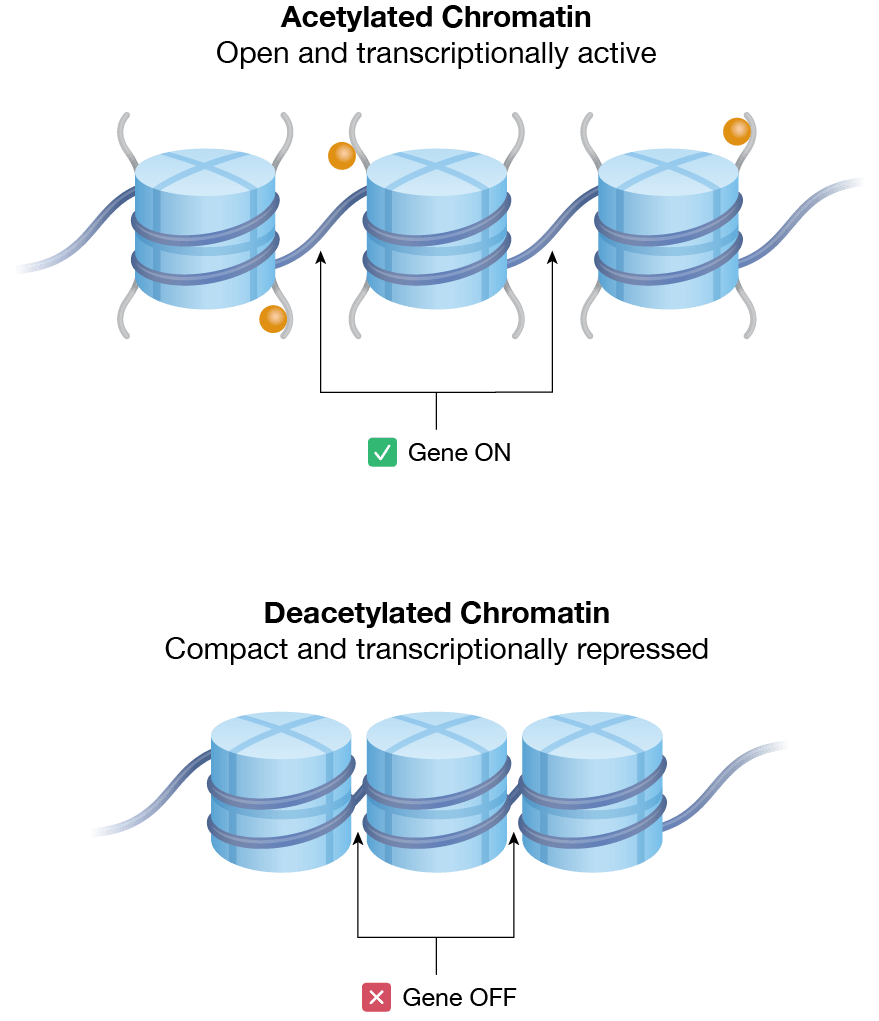
Figure 1. Chromatin organization and epigenetic regulation. a) Nucleosomes are the most basic level of chromatin organization; they consist of DNA wrapped around histone proteins. The histone proteins of each nucleosome assemble as an octamer, also known as histone octamer. The cell regulates access to specific genes by adding or removing post-translational labels on the histone tails (orange). Direct methylation of DNA (pink) can also regulate gene expression. b) Relaxed chromatin is “open” or transcriptionally active. Acetylation of histone tails (orange) is associated with a transcriptionally active chromatin state. Conversely, densely packed chromatin is transcriptionally repressed. Deacetylation of histone tails is associated with transcription repression.
Chromatin Structure
Inside the nucleus, DNA is spooled around a class of structural and regulatory proteins called histones (Figure 1a). Each histone is organized as an octamer; two copies of four different histone proteins. The individual units of DNA and histone are called nucleosomes. Nucleosomes are connected via stretches of “linker” DNA like beads on a string. The highly ordered packaging of DNA and histones together is called chromatin.
Chromatin packing influences the accessibility of promoter regions to transcription factors and represents a major means of epigenetic gene regulation (Figure 1b). Relaxed or “open” chromatin, also known as euchromatin, allows transcription factors more access to promoter sites, thereby facilitating transcriptional activation. In contrast, more densely packed heterochromatin conceals DNA promoter sequences from transcription factors. Epigenetic modifications to the histone proteins and DNA modulate the affinity of chromatin-binding proteins, in turn altering chromatin structure.
Histone Modification
Histones are the major protein component of chromatin and are subject to several different post-translational modifications, including methylation, acetylation, phosphorylation, ubiquitylation, and SUMOylation. These modifications, often in concert, occur on the highly basic histone tails and play a critical role in chromatin packing.
Histone tails are positively charged and interact with both negatively charged DNA and acidic patches of nucleosomes. Histone acetyltransferases (HATs) are a class of enzymes that modulate these interactions to prompt the formation of euchromatin from heterochromatin. HATs catalyze the transfer of an acetyl group to conserved lysine residues on the histone tail, promoting a relaxed (transcriptionally active) chromatin. In contrast, histone deacetylases (HDACs) catalyze the removal of acetyl groups from histones, leading to more tightly packaged (transcriptionally inactive) chromatin.
The recruitment of HATs and HDACs is tightly regulated by DNA binding proteins located near epigenetic target regions. Examination of histone acetylation patterns has demonstrated a high correlation between histone acetylation and active transcription, whereas histone methylation can be associated with the activation or silencing of genes depending on the amino acid modified and the number of methyl groups added. The concept of multiple dynamic modifications regulating gene expression in a systematic and reproducible fashion is known as the histone code.
Chromosomal DNA Modifications
In both prokaryotes and eukaryotes, gene expression is also regulated by DNA methylation, the addition of methyl groups to DNA without affecting the coding sequence. DNA methyltransferases (DMNTs), a conserved family of cytosine methylases, selectively methylate the 5'-position of cytosines in the promoter and coding region of genes. DNA methylation leads to an additional level of transcriptional repression via the recruitment of enzymes that methylate histones and promote a heterochromatin state. This link between DNA methylation and histone methylation represents a synergistic repression of gene expression.
In contrast to methylcytosine modifications, hydroxymethylcytosines catalyze the formation of 5'-hydroxymethylcytosines in gene promoter regions and are often associated with open chromatin (Ficz 2011). Differential expression of methylcytosine and hydroxymethylcytosine DNA modifications likely plays a significant role in dictating gene expression patterns throughout cell development.
Methods Used to Study Epigenetics
Epigenetics analysis techniques are used to assess methylation patterns, DNA/protein interactions, and chromatin accessibility (Hamamoto 2019). Prior to next generation sequencing (NGS), chromatin immunoprecipitation (ChIP) assay and bisulfite sequencing (BS-Seq) in combination with microarrays were the dominant methodologies. The advent of NGS made genome-wide epigenetics analysis possible for a range of species and cell types, enriching public databases like EpiFactors, MethDB, and dbEM.
DNA Methylation Analysis
In mammals, DNA methylation is observed on 80% of cytosines in cytosine/guanine (CpG) regions and has published links to cancer. Bisulfite sequencing (Barros-Silva 2018) is commonly used to identify individual methylation sites and quantify the level of methylation in a specific region. This protocol converts unmethylated cytosine residues to uracil, while methylated cytosines remain unmodified and are analyzed through downstream PCR, qPCR, and sequencing steps. Post NGS, reduced representation bisulfite sequencing (RRBS) was developed as a high-throughput technique to analyze genome-wide methylation profiles.
High resolution melt (HRM) analysis is the qualitative analysis of a DNA fragment's melt curve following PCR amplification. It is a simple, cost-effective, and rapid tool for studying genetic variation, with applications in single nucleotide polymorphism (SNP) analysis, mutation scanning, species identification, and methylation analysis. In this method, DNA is first treated with sodium bisulfite to convert unmethylated cytosine residues to uracil, followed by PCR amplification and melt curve analysis. The varied melt temperature from PCR products originating from bisulfite-treated DNA is indicative of DNA methylation status. Robust HRM analysis requires a real-time PCR (qPCR) detection system with thermal stability, sensitivity and dedicated software, such as the CFX qPCR series. For more details, see What Is High-Resolution Melting?
Methylated DNA immunoprecipitation (MeDIP) is used to examine genome-wide changes in DNA methylation patterns across varied cell types. In this technique, DNA is isolated from cells and sheared using sonication. Antibodies specifically targeting methylated DNA fragments are used to isolate methylated regions. The methylated DNA fragments can be identified using high-resolution DNA microarray or NGS techniques.
Analysis of DNA/Protein Interactions
Histone modifications like phosphorylation, methylation, and ubiquitination are mapped using chromatin immunoprecipitation (ChIP). Here, DNA-binding proteins are first isolated using highly specific antibodies and then analyzed by nucleic acid techniques like PCR, qPCR, microarray hybridization, and sequencing. The combination of ChIP and NGS is used for genome-wide analysis of histone modifications and transcription factor binding.
Chromatin Accessibility
Native chromatin states can be experimentally identified using assays that interrogate nuclease accessibility of chromatin. Nucleases are enzymes that digest nucleic acids into their monomer constituents. However, nucleases are unable to digest closed, inaccessible chromatin, leaving the DNA intact for subsequent examination.
Digital DNase and DNase-seq are two methods that employ nuclease digestion to generate short DNA strands from nuclease-accessible genomic regions. These short DNA strands are isolated and examined by NGS to provide detailed sequence information on genome stretches accessible to nuclease digestion. While DNase-seq is useful for detecting the location of regulatory proteins in the genome, it has limitations in sensitivity and an inherent bias due to the sequence-specificity of DNase I. Researchers use these techniques to compare DNA-structural differences between samples on a genome-wide scale and understand how observed differences lead to disease.
A faster assay for whole genome chromatin accessibility studies, is transposase accessible chromatin (ATAC-seq). In the ATAC-seq method, adapters for high-throughput sequencing are ligated into accessible regions using the action of a hyperactive Tn5 transposase and mapped by sequencing (Figure 2). One unique advantage of ATAC-seq is that it is compatible with a lower number of cells, which is important when processing precious samples like patient tumor cells. Further, unlike ChIP, this method does not require fixing of the sample, so the collected data reflects the native chromatic state.
Simplified Workflow for ATAC-seq

Figure 2. ATAC-seq Workflow. The ATAC-seq assay is used to detect chromatin accessibility. In this assay, hyperactive Tn5 transposase can only access and modify DNA regions in an “open” chromatin state. The enzyme inserts sequencing adapters (or priming sequences) into open regions through a process called tagmentation. The resulting DNA fragments are then amplified and sequenced via high throughput sequencing.
Chromatin Conformation Assays
Spatial changes in the nuclear localization of genomic regions are thought to influence gene expression. Chromatin looping, for example, brings sites from different chromosome locations into proximity and facilitates the transfer of RNA polymerase between genomic regions. The chromosome conformation capture (3C) assay is used to examine chromatin looping. In this assay, chromatin is crosslinked prior to cell lysis, and then exposed to restriction digest. Gene-specific PCR primers are then used to examine looping of genomic regions that are hypothesized to be in close proximity (Gavrilov 2009).
Modified versions of the 3C assay have been developed for other epigenetic analyses. The circular chromosome conformation capture (4C) enables researchers to assess interactions between genetic loci in a high-throughput fashion (Ohlsson 2007). Chromosome Conformation Capture Carbon Copy (5C) also provides a high-throughput analysis of genomic interactions by using NGS technologies to identify novel DNA regions in close proximity within the cell (Dostie 2006).
Single-Cell Epigenomics
A new generation of single-cell epigenomics tools are helping us understand the cell-to-cell variation in gene regulation and how they manifest in development and disease (Shema 2019). Single-cell methods involve a cell isolation step using a range of methods, including microfluidics such as flow sorting or droplet capture. DNA methylation on specific nucleotides is profiled using single-cell RRBS or the whole genome version (WGBS). Single-cell ChIP-seq utilizes droplet microfluidics to isolate cells and study histone modifications across the genome. To study spatial organization of chromatin in single cells, a modified version of the 3C assay called Hi-C is used.
Single-cell DNA-seq and ATAC-seq use fluorescence cell sorting or microfluidics to isolate cells and profile chromatin accessibility. Adapted ATAC-seq methods utilizing barcodes and combinatorial indexing allow for processing of tens of thousands of cells in parallel (Cusanovich, 2015; Buenrostro 2015). Advancements in the field have led to protocols and devices for performing single-cell ATAC-seq assays with applications in biomarker discovery, gene expression regulation and cellular heterogeneity studies (Figure 3).
Simplified Workflow for Single-Cell ATAC-seq
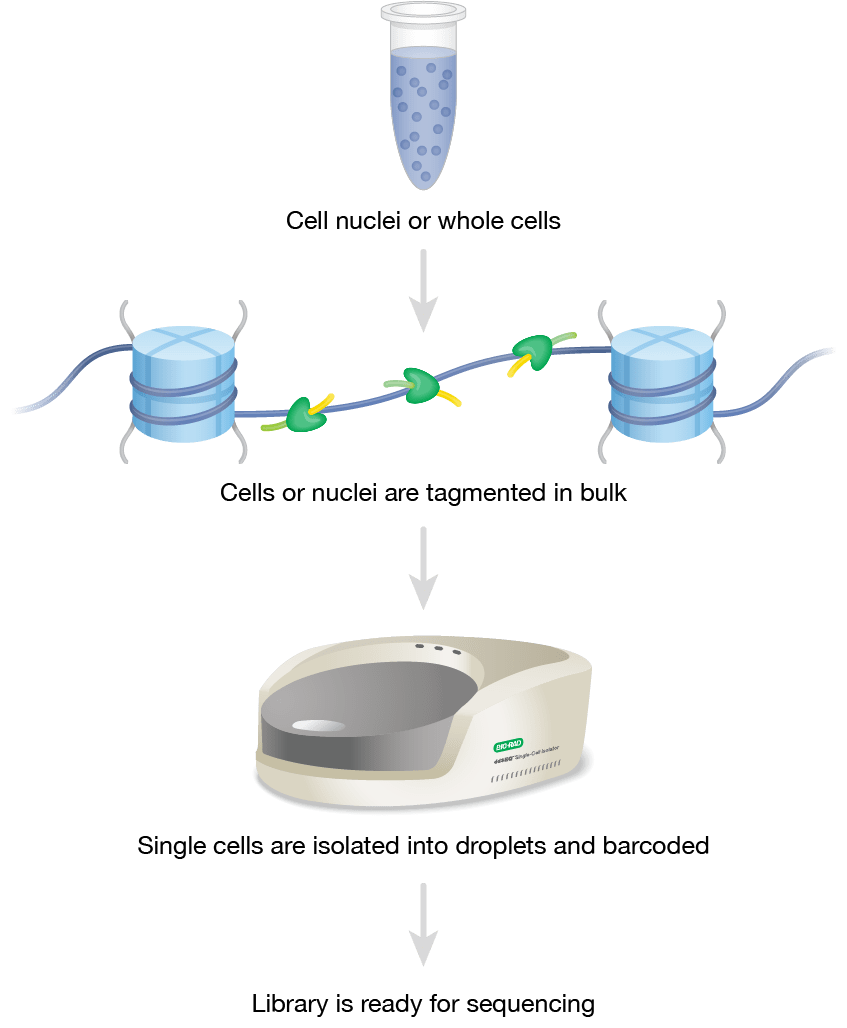
Figure 3. ddSEQ Single-Cell ATAC-seq Workflow. Single-cell ATAC-seq allows for investigation of open chromatin on a cell-to-cell basis. This protocol uses cell nuclei or whole cells as start material. Next, hyperactive Tn5 transposase cuts and ligates sequencing adapters into open chromatin regions in bulk (tagmentation). Then, single cells, barcodes and sequencing reaction components are isolated into droplets by an instrument like the ddSEQ Single-Cell Isolator. The resulting library is sequenced on a high-throughput NGS sequencing platform.
Learn More about the ddSEQ Single-Cell ATAC-Seq Library Prep Kit »
Clinical Implications of Epigenetics
Epigenetics is an emerging field with important implications in medicine. Modifications, like DNA methylation, regulate chromatin structure and influence gene expression, with documented links to diseases (Jin 2018) like cancer, autoimmune disease, and metabolic disorders, as well as processes like cellular aging (Ciccarone 2018). Biomedical research is exploring how epigenetic regulation can be exploited for the development of novel drug therapies and biomarkers for disease (Laird 2010).
Environmental Factors in Epigenetics
Environmental factors like stress, nutrition, exercise and drugs can influence the dynamics of epigenomics. Dietary folate for example can affect methylation and dietary deficiencies have been linked to colorectal cancer via genomic hypermethylation. Stress in utero and during childhood has also been linked to differential DNA methylation and varied gene expression patterns.
Epigenetic Changes in Cancer
The role of epigenetics in cancer has been widely reported. Methylation has been linked to DNA repair, metastasis, angiogenesis, drug resistance, and other processes involved in cancer biology. Hypermethylation of the tumor suppressor protein TP53, for example, is a confirmed cancer biomarker, and both hyper- and hypo-methylation of genes related to breast cancer have been suggested for early detection. Histone modification is also known to promote cancer, including lung, prostate, colorectal, and pancreatic cancers. In colorectal cancer, non-coding RNAs, which also act epigenetically to regulate gene expression, are shown to play a role in many involved pathways (Kita 2017).
Autoimmune Disease
Epigenetic regulation is one of the documented factors that influences disease expression in some autoimmune/inflammatory diseases. Down regulation of histone proteins, histone modifying genes, and genes associated with DNA methylation, for example, are observed in biopsy samples of patients with monogenic autoinflammatory disorder cryopyrin-associated periodic syndromes (CAPS) (Aubert 2012). Studies have also shown epigenetic regulation in the pathophysiology of autoinflammatory bone condition chronic nonbacterial osteomyelitis (Hofmann 2012).
Neurodegeneration and Epigenetics
Epigenetic regulation plays an important role in learning and memory in the adult brain. Evidence also suggests a link between epigenetics and neurodegenerative disorders. Histone modification for example, plays a role in neural cell death, which causes memory loss. A 2012 study in a model of ischemic stroke showed that repressor element-1 silencing transcription factor (REST) induced epigenetic remodeling and silencing of target genes. Importantly, blocking REST activity prevented the observed remodeling events and promoted neuronal cell survival (Noh 2012).
Role of Epigenetics in Stem Cell Differentiation
Studies of the epigenetic changes that occur during stem cell differentiation have provided clues as to how the epigenetic status of cells may impact pluripotency (Atlasi 2017). A 2004 study showed that histone acetylation was required in the in vitro differentiation of murine embryonic stem cells (Lee 2004). Epigenetic manipulations may enable researchers to more accurately direct differentiation of cells into desired cell types and may aid in the generation of induced pluripotent stem cells.
Epigenetics in Infectious Disease Processes
Epigenetics regulate how the immune system responds to infectious disease (Cole 2016). Macrophages, for example, have shown varied patterns of histone modification and gene transcription activity depending on their activation state (Ehrt 2001 and Satoh 2010). A 2020 study has revealed how epigenetics can promote drug resistance mechanisms in microbes. In the study, researchers observed heterochromatic-mediate gene silencing in yeast that were resistant to an antifungal agent (Torres-Garcia 2020).
Related Topics and Products
Discover lncRNA: Understanding the Long Noncoding Transcriptome
Learn about the exciting new field of long noncoding RNA (lncRNA) and how this newly discovered class of RNA compares to messenger RNA (mRNA).

Advances in RNAseq
NGS RNA sequencing (RNA-Seq) is the foremost tool for transcriptomics, gene expression profiling, and the study of differential RNA splicing.

RNA-Seq Workflow
A guide to the steps of an RNA-Seq workflow including library prep and quantitation and software tools for RNA-Seq data analysis.

PCR (Polymerase Chain Reaction)
Learn how PCR works, how to choose the right PCR instrument, how to design, optimize, analyze, and troubleshoot PCR assays.

Real-Time PCR (qPCR)
Discover an array of solutions to optimize, execute, and troubleshoot MIQE-compliant real-time PCR experiments.

Digital PCR (ddPCR)
Learn how breakthrough digital PCR technology is used in droplet digital PCR (ddPCR) to provide ultrasensitive and absolute nucleic acid quantitation.

lncRNA RT-qPCR Workflow
Bio-Rad's innovative workflow enables highly sensitive detection of long noncoding RNA (lncRNA) and helps to overcome challenges associated with IncRNA expression analysis.
Next-Generation Sequencing
Novel products for NGS library preparation and quantitation used in cutting-edge applications such as whole-transcriptome RNA-Seq and single-cell sequencing.

Traditional PCR Systems
DNA amplification instruments range from personal thermal cyclers to the flexible 1000-series.

qPCR Detection Systems
These systems deliver sensitive, reliable detection of both singleplex and multiplex real-time PCR reactions.

PCR & qPCR Reagents
A wide range of reagents for reverse transcription, PCR, and real-time PCR, optimized to generate accurate and reproducible data.

PCR Plastic Consumables
A large selection of thin-wall PCR tubes, plates, seals, and accessories precisely manufactured for optimal fit and cycling performance in Bio-Rad thermal cyclers, real-time PCR systems, ddPCR systems, and all major competitor systems.

PrimePCR PCR Primers, Assays & Arrays
Experimentally validated PCR primer and probe assays for gene expression, copy number variation, and mutation detection analysis for real-time PCR and Droplet Digital PCR.
Droplet Digital PCR Assays
Bio-Rad offers a comprehensive portfolio of Digital PCR Assays and Kits for numerous applications including Mutation Detection, Copy Number Determination, Genome Edit Detection, Gene Expression, Expert Design Assays, Residual DNA Quantification and Library Quantification.
References
- Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet. 2017 Nov;18(11):643-658. doi: 10.1038/nrg.2017.57. Epub 2017 Aug 14. PMID: 28804139.
- Aubert P, Suárez-Fariñas M, Mitsui H, Johnson-Huang LM, Harden JL, Pierson KC, Dolan JG, Novitskaya I, Coats I, Estes J, Cowen EW, Plass N, Lee CC, Sun HW, Lowes MA, Goldbach-Mansky R. Homeostatic tissue responses in skin biopsies from NOMID patients with constitutive overproduction of IL-1β. PLoS One. 2012;7(11):e49408. doi: 10.1371/journal.pone.0049408. Epub 2012 Nov 30. PMID: 23226210; PMCID: PMC3511496.
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015 Jul 23;523(7561):486-90. doi: 10.1038/nature14590. Epub 2015 Jun 17. PMID: 26083756; PMCID: PMC4685948.
- Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev. 2018 Sep;174:3-17. PMID:29268958.
- Cole J, Morris P, Dickman MJ, Dockrell DH. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharmacol Ther. 2016 Nov;167:85-99. doi: 10.1016/j.pharmthera.2016.07.013. Epub 2016 Aug 9. PMID: 27519803; PMCID: PMC5109899.
- Cusanovich, D. A. et al. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015).
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006 Oct;16(10):1299-309. doi: 10.1101/gr.5571506. Epub 2006 Sep 5. PMID: 16954542; PMCID: PMC1581439.
- Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G, Nathan C. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001 Oct 15;194(8):1123-40. doi: 10.1084/jem.194.8.1123. PMID: 11602641; PMCID: PMC2193509.
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011 May 19;473(7347):398-402. doi: 10.1038/nature10008. Epub 2011 Apr 3. PMID: 21460836.
- Gavrilov A, Eivazova E, Priozhkova I, Lipinski M, Razin S, Vassetzky Y. Chromosome conformation capture (from 3C to 5C) and its ChIP-based modification. Methods Mol Biol. 2009;567:171-88. doi: 10.1007/978-1-60327-414-2_12. Erratum in: Methods Mol Biol. 2009;567:E1. PMID: 19588093.
- Hamamoto R, Komatsu M, Takasawa K, Asada K, Kaneko S. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules. 2019 Dec 30;10(1):62. doi: 10.3390/biom10010062. PMID: 31905969; PMCID: PMC7023005.
- Hofmann SR, Morbach H, Schwarz T, Rösen-Wolff A, Girschick HJ, Hedrich CM. Attenuated TLR4/MAPK signaling in monocytes from patients with CRMO results in impaired IL-10 expression. Clin Immunol. 2012 Oct;145(1):69-76. doi: 10.1016/j.clim.2012.07.012. Epub 2012 Aug 4. PMID: 22940633.
- Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5(1):1–8. Barros:10.1016/j.gendis.2018.01.002.
- Kita Y, Yonemori K, Osako Y, Baba K, Mori S, Maemura K, Natsugoe S. Noncoding RNA and colorectal cancer: its epigenetic role. J Hum Genet. 2017 Jan;62(1):41-47. doi: 10.1038/jhg.2016.66. Epub 2016 Jun 9. PMID: 27278790.
- Laird PW (2010). Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 3, 191–203. PMID: 20125086
- Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004 Jan;38(1):32-8. doi: 10.1002/gene.10250. PMID: 14755802.
- Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012 Apr 17;109(16):E962-71. doi: 10.1073/pnas.1121568109. Epub 2012 Feb 27. PMID: 22371606; PMCID: PMC3341013.
- Ohlsson R, Göndör A. The 4C technique: the 'Rosetta stone' for genome biology in 3D? Curr Opin Cell Biol. 2007 Jun;19(3):321-5. doi: 10.1016/j.ceb.2007.04.008. Epub 2007 Apr 26. PMID: 17466501.
- Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010 Oct;11(10):936-44. doi: 10.1038/ni.1920. Epub 2010 Aug 22. PMID: 20729857.
- Shema E, Bernstein BE, Buenrostro JD. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat Genet. 2019 Jan;51(1):19-25. doi: 10.1038/s41588-018-0290-x. Epub 2018 Dec 17. PMID: 30559489.
- Surace AEA, Hedrich CM. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front Immunol. 2019 Jul 4;10:1525. doi: 10.3389/fimmu.2019.01525. PMID: 31333659; PMCID: PMC6620790.
- Torres-Garcia S, Yaseen I, Shukla M, Audergon PNCB, White SA, Pidoux AL, Allshire RC. Epigenetic gene silencing by heterochromatin primes fungal resistance. Nature. 2020 Sep;585(7825):453-458. doi: 10.1038/s41586-020-2706-x. Epub 2020 Sep 9. PMID: 32908306; PMCID: PMC7116710.