


Discover how electroporation can be used to generate transgenic zebrafish. This foundational study by Buono and Linser describes how the Bio-Rad Gene Pulser™ System can be used to introduce plasmid DNA into Brachydanio rerio embryos via electroporation. You’ll see how applying electroporation at early developmental stages supports embryo survival and transgene expression, providing a higher-throughput alternative to microinjection. Read on to explore the full study
Featured Content
Russell J. Buono and Paul J. Linser, Whitney Marine Laboratory, University of Florida, 9505 Ocean Shore Blvd., St. Augustine, Florida 32086, 904-461-4000
Introduction
The production of transgenic animals has proven to be a powerful tool in the study of developmentally regulated genes. The traditional method of microinjection of foreign DNA into fertilized embryos is tedious and time consuming. We have used the Bio-Rad Gene Pulser® apparatus to generate transgenic fish by electroporation. To establish our technique we used the plasmid construct RSVCAT (Gorman et al., Proc. Natl. Acad. Sci. USA, 79, 6777-6781, 1982). This plasmid uses the Rous sarcoma virus (RSV) promoter to effectively drive expression of chloramphenicol acetyl transferase (CAT) in numerous eukaryotic cell types.
Methods
Fish Maintenance and Egg Collection
Zebrafish (Brachydanio rerio, Figure 1) are obtained from the local pet store and maintained in a 20 gallon aquarium in deionized water (dH20) at 26° C. Fish are fed three times daily: flake food in the morning and at noon, and live hatchling artemia in the afternoon. Fish spawning is induced by the onset of light. Ten to twelve fish are placed in a net breeder with a 1/16th inch nylon mesh net. The net breeder is placed in a 5 gallon aquarium (used only for breeding) with dH20 maintained at 27° C and suspended over a plexiglass container to collect spawned eggs. All tanks are in a room with windows and receive natural sunlight. The fish spawn at first light and eggs are fertilized externally. Fertilized eggs are collected soon after spawning; most embryos are at the two- to four-cell stage. The eggs are rinsed in a fine mesh dip net with dH20 and transferred to a 100 mm petri dish with 10 ml dH20. Embryos are washed in 0.1% (v/v) surgical scrub (Wescodyne) for 5 minutes while debris is physically separated using a plastic transfer pipet. The embryos are then washed twice with fresh water.

Plasmid Preparation
The plasmid RSVCAT is purified by cesium chloride density gradient ultracentrifugation. DNA is suspended in TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and 100 µg are digested overnight with BamHI at 37° C. The digest is extracted once with phenol/chloroform and once with chloroform alone. The DNA is precipitated from the aqueous phase using absolute ethanol. This DNA is then resuspended in calcium-free phosphate buffered saline (PBS/-Ca++) at a concentration of approximately 100 µg/ml and is used several times for electroporation.
Electroporation
Up to two hundred embryos are rinsed in PBS/-Ca++ and placed in a 0.4 cm wide cuvette with 800 µl of the DNA solution. The embryos are pulsed three times using the 0.25 µF capacitor of the Gene Pulser apparatus (Bio-Rad). The pulse field strength is 125 V/cm, and each time constant is approximately 7-10 msec with a pulse interval of 1 sec. The DNA solution is removed, and the embryos are rinsed in dH20 and then placed into a 100 mm petri dish in fresh dH20 at 26° C. Sham electroporation is done with PBS/-Ca++ alone.
Screening for the Transgene
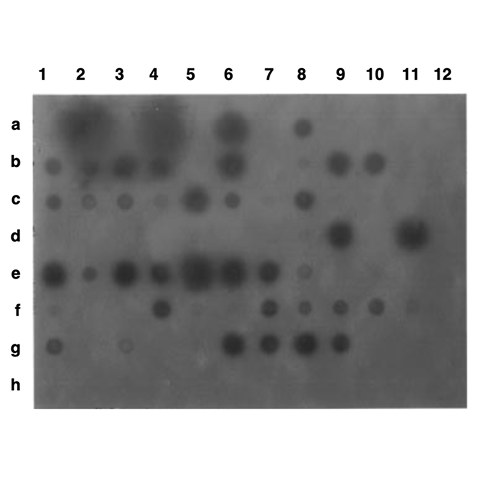
Zebrafish embryos hatch 72 to 96 hours after spawning, and development appears morphologically normal. Six days after spawning a random sample of fry are sacrificed and digested with proteinase K overnight at room temperature. The proteins are removed by phenol/chloroform extractions and the genomic DNA is precipitated from the aqueous phase using absolute ethanol. The DNA is resuspended in 40 µl of TE, half of which is used for a dot blot. DNA for the dot blot is alkaline denatured, vacuum blotted onto nitrocellulose membrane, and covalently linked to the membrane by ultraviolet irradiation. Prehybridization is with 0.05x BLOTTO (Sambrook, J., Fritsch, E. F., and Maniatas, T., Molecular Cloning, 2nd edition, vol. 2, chapter 9, 1989) in 6x SSC for 2 hours at 68° C. The CAT gene (1.6 kb) is cut out of a plasmid vector and the gel-purified linear fragment is used as a probe. The probe is labeled by random priming and 1 x 105 to 1 x 106 cpm are added per milliliter to the prehybridization solution. Hybridization takes place overnight at 68° C. Dot blots are washed the following morning, covered with plastic wrap, and placed on X-ray film.
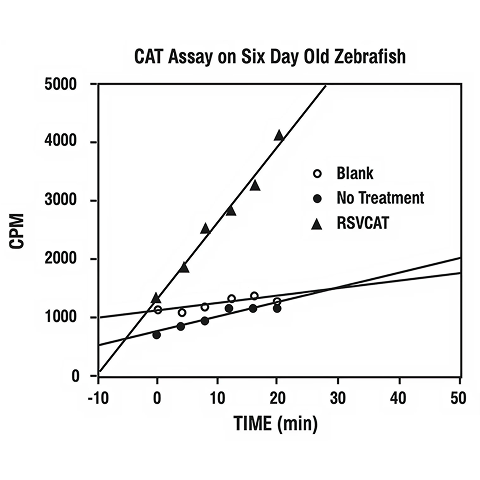
In one instance 25 possible transgenic fish (6 days posthatch) were homogenized in 100 µl of 0.25 M Tris-HCl, pH 7.6, and the soluble proteins tested for CAT enzyme activity (Neumann, J. R., et al., BioTechniques, 5, 444-447, 1987). Nontreated fish were used as controls.
Southern blots are in progress at the time of this writing to determine integration of the transgene into the host genome.
Table 1
| Embryos Electroporated | # Survivors 2 Days Posthatch | % Survivors | # Tested by Dot Blot | # Positive | % Positive | |
|---|---|---|---|---|---|---|
| RSVCAT | 930 | 639 | 68% | 210 | 137* | 65% |
| Sham | 130 | 74 | 57% | 15 | 0 | 0% |
| No Treatment | 115 | 105 | 91% | 15 | 0 | 0% |
| *Seventy of these positives generated a signal as strong or stronger than the 60 pg control, i.e., 30% strong positives. | ||||||
Results
The data from three separate experiments are totaled and shown in Table 1. The results indicate that over 50% of the animals survive the electroporation conditions, and that over 50% of the animals tested carry the transgene to some extent (based on the dot blot analysis). Of these positive animals, 30% show strong hybridization to the probe (signal as strong or stronger than the 60 pg control). A typical dot blot is shown in Figure 2.
Figure 3 shows the results of a CAT enzyme assay of the soluble proteins of 25 possible transgenic fish (6 days posthatch) compared to that activity found in the proteins of 25 non-treated fish. This graph shows that the transgene is being transcribed and translated into functional CAT enzyme.
Discussion and Conclusion
The generation of transgenic fish by electroporation has several advantages over the traditional microinjection technique. The procedure is rapid and simple, and it allows treatment of many embryos in a short period of time.
However, our claims for the technique are not without some caveats. At this point, we can not assert that any animals which carry large amounts of foreign DNA (determined by dot blot analysis) have integrated that DNA into the host genome. Southern blot analysis is needed to show integration. Though it is unlikely that plasmid DNA (most in the linear form) could be carried over in the animals' cells through many rounds of mitotic division, it is a possibility. Future experiments are planned using gel-purified linear fragments of various constructs to try to avoid supercoiled plasmid carryover.
Also, there is great variability in the amount of signal seen from one positive animal to the next. This may represent non-integration or low numbers of cells carrying copies of the transgene. It is therefore prudent to electroporate only one- and two-cell embryos to avoid generating chimeric or mosaic animals.
We have not shown stable integration of the transgene. Stable integration can only be demonstrated by the production of transgenic progeny from transgenic founder animals. This point may not be crucial to our studies since we are interested in tissue- and cell-specific promoter function during development. Hence, our animals, which carry the transgene during maturation, may be sufficient to answer our immediate questions. Nonetheless production of stable lines of transgenic animals will be necessary for future investigations. It has been shown that foreign DNA introduced into zebrafish embryos is degraded over time, resulting in mosaic animals, some of which can give rise to stable lines of transgenic fish (Stuart, G. W., McMurray, J. V., and Westerfield, M., Development, 103, 403-412, 1988). We are currently raising possible transgenic hatchlings to test transmission of foreign DNA to progeny.
We have shown that electroporation may be a viable alternative to microinjection. It is clear that our treated animals are carrying the foreign DNA, some in large amounts, and that foreign DNA is being transcribed and translated into protein. Survival rates and numbers of animals testing positive for the transgene are very high. It is evident that additional work needs to be done on this system, however, we believe our initial experiments are very encouraging. We intend to eliminate the problems we have discussed and make the production of transgenic zebrafish by electroporation a standard technique.
This work was supported by a basic research grant (#1-1003) from the March of Dimes Birth Defect Foundation.
Featured Content
Transfection Solutions

Gene Pulser Xcell Electroporation System
Tried and trusted by thousands of researchers worldwide, transfect every cell type from primary, suspension, and difficult to transfect cells with the Gene Pulser Xcell Electroporation System.

Questions about transfection?