
The rapid advancement of next generation sequencing (NGS) technologies over the last two decades has revolutionized the field of genomics and made sequencing accessible to many research and clinical applications (Goodwin 2016; Stark 2019). Established and new market players are now focused on pushing the limits of speed and cost, helping establish sequencing as a routine part of clinical care. Moreover, newer generations of instruments continue to make gains in areas of long-range, direct, and single-cell sequencing, in addition to introducing novel types of measurement. Here, we highlight a handful of technologies and applications that are at the forefront of these efforts.
Future-Generation Sequencing
While standard short-read sequencing technologies dominate the current NGS market, newer third-generation instruments have opened the door to long-read, direct-read, and single-cell sequencing (Goodwin 2016; Stark 2019). These technologies address some of the key limitations of short-read sequencing and are becoming more prevalent in the field of medical genetics.
Long-Read Sequencing
In contrast to short-read protocols that read a few hundred nucleotides at a time, long-read sequencing technologies can read up to 200 kb of DNA in a continuous fashion. This difference in read length is significant when studying genomic regions with high GC content, repetition, and structural variations. These regions pose a challenge when assembling sequences from short reads, even with the most sophisticated computational algorithms. Importantly, the lack of a PCR step in contemporary protocols reduces coverage bias of GC-heavy regions, some of which may be implicated in human diseases.
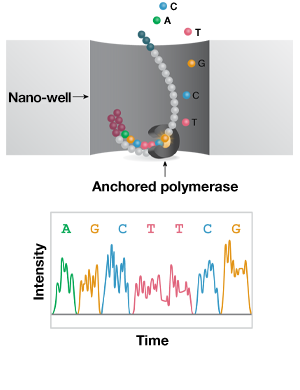
The key technologies in this space are single-molecule real-time (SMRT) sequencing and nanopore sequencing. In SMRT sequencing, the sequencing-by-synthesis reaction takes place at the bottom of nanowells, where individual polymerase molecules process one molecule of template, per well. Nanopore sequencing, utilizes a helicase-pore complex to couple each nucleotide to a defined shift in electrical current as it passes through; this allows “direct” sequencing of the DNA or RNA template. These “physical” approaches differ from synthetic long reads that computationally reconstruct sequences from short-read data collected on barcoded fragments.
Long-read sequencing is a powerful tool for genomics and transcriptomics studies and a complement to existing short-read methodologies (Mantere 2019). These technologies have been used to resolve allele phasing, discriminate pseudogenes, map structural variants, and sequence tandem repeat regions, all of which can be important in the etiology of human diseases, as shown in a recent allele phasing study involving cystic fibrosis patients (Regan 2015). In cancer research, long-read sequencing is capable of genotyping complicated cancer genomes, including structural variants and short indels. Improvements in accuracy, cost, and data analysis will further integrate long-read sequencing in diagnosis, treatment planning, and other facets of clinical care.
A. Sequencing by synthesis (Short Read)

B. Single-molecule real-time (Long Read)
C. Nanopore (Direct Long-Read)

Short-read vs. long-read sequencing technologies. A. Template DNA is amplified by bridge PCR to generate clusters on a flow cell. In each reaction cycle, fluorescent nucleotides are incorporated into the growing strand, generating a color signal. B. In single-molecule real-time (SMRT) long-read sequencing, individual molecules of adapter-ligated template are bound to a single polymerase molecule in a nanowell. During DNA polymerization of the complementary strand, the wells are imaged to identify the incorporated fluorescent nucleotide. C. In nanopore single-molecule long-read sequencing, the template is ligated to a motor protein and delivered to a flow cell. Here, the motor protein threads the template through a nanopore, where a change in current is associated with specific nucleotides.
Learn more about Next-Generation Sequencing Technologies »
Single-Cell Sequencing
Traditional sequencing protocols performed on templates extracted from cell mixtures do not capture the genomic variation of individual cells. Single-cell sequencing of DNA or RNA captures these differences and is used to investigate new questions in biology. Single-cell methods first require the isolation of single cells by various means including physical manipulation such as micropipetting in tubes, microfluidic methods such as flow sorting or droplet capture, applied electric fields, and optical techniques such as optical tweezers, with distribution into nanowells and in situ barcoding of cells (Shinde 2018). Following library generation, the template is sequenced on an NGS platform.
In addition to other third-generation NGS methods, single-cell DNA sequencing is emerging as a powerful tool in studies of the microbiome. A growing number of studies have linked the human microbiome to overall wellness and a range of diseases including cancer and have ushered in novel treatments centered on modifying the gut microbiome (Bik 2016). Amplicon and metagenomics sequencing approaches have been widely used in taxonomic and functional studies of the microbiome, but these methods are limited in resolution and coverage, respectively (Cheng 2019). Using single-cell sequencing, scientists can obtain high-quality data from low-abundance species, or species with unknown cultivation conditions, and determine functional links at the strain level. Single-cell technologies combined with metagenomics approaches will shed critical insights into the relationship between the gut microbiota and major organs like the brain. Importantly, emerging data on virus-host interactions and pathogenic microbes will inform on the pathogenesis and treatment of infectious diseases.
In transcriptomics, single-cell RNA sequencing (scRNA-Seq) has been used to study gene interaction networks, lineage tracing, and rare-cell populations, with applications in immunology and tumor heterogeneity (Hwang 2018; Stark 2019). Large atlas projects using scRNA-Seq aim to map the 35 trillion cells of the human body (Human Cell Atlas), and with reduced sequencing cost and better bioinformatics capabilities, will shed new insights into the complexity of multicellular biological systems.

Single-cell sequencing by droplet capture. This technology utilizes disposable microfluidic cartridges to coencapsulate single cells and barcodes into subnanoliter droplets, where cell lysis and barcoding occur. RNA-Seq libraries are subsequently prepared and sequenced.
Spatially Resolved Sequencing
While standard bulk and single-cell sequencing data is disconnected from cellular context, spatialomics preserves spatial information and allows for a better understanding of complex diseases (Burgess 2019). As an example, in a recent study, gene expression profiles were mapped across prostate cancer tissue samples, revealing “high-risk” cancer-prone areas beyond the annotated tumor boundaries (Berglund 2018). In transcriptomics studies, spatial encoding and in situ transcriptomics capture spatial data prior to sequencing using either laser capture micro-dissection or barcoding of RNAs prior to isolation. These methods have been successfully used on tissue sections from a variety of species and report abundances with more sensitivity than traditional fluorescence in situ hybridization (FISH)-based methods. The most recent technology, high-throughput fluorescent in situ sequencing method (FISSEQ) allows in situ RNA sequencing directly within tissue, cell culture, and whole-mount embryos.
Clinical Opportunities
Point-of-Care Sequencing
NGS is an invaluable clinical tool for identifying genetic factors that cause disease or determine response to a given treatment (Pereira 2017). Multi-gene panels are being considered as a cost-effective approach for identifying the genetic basis of complex diseases and cancers of unknown origin. Whole-genome and whole-exome sequencing can potentially reveal rare genetic disorders and cancer risk factors, but due to their high costs and potential for discoveries without clear clinical meaning, these methods are employed on a case-by-case basis. Clinical oncologists can now use RNA sequencing for cancer typing and profiling tumor and immune checkpoint markers to assess treatment response, while newer protocols are being applied to circulating tumor DNA and RNA (ctDNA and ctRNA) discovery. While some diagnostic genetic tests (e.g., prenatal testing) are already approved for use, wide adoption of genetic testing in the clinic faces many challenges. Data reliability, data interpretation, quality control, and best practices for communicating results with patients are among the important questions that must be addressed.
Learn more about how integrating ddPCR technology in your NGS workflow improves your research »
Rare Disease Diagnosis
The diagnosis of rare genetic diseases (defined as < 1/15,000 in the US) has historically been a long, often watch-and-wait process, with late access to sequencing information (Liu 2019). NGS has dramatically changed this workflow by allowing genomic testing early on, providing significant benefits to patients. Over the last decade, whole-exome and genome sequencing have helped identify disease-causing genes in rare metabolic, neurodevelopmental, neuromuscular, movement, and demyelinating disorders (Fernandez-Marmiesse 2018). Of great interest are gene panels that focus on the coding regions of relevant genes as a cost-effective and practical NGS-based diagnostic tool for rare diseases. As with most clinical tools, accuracy and data interpretation is an ongoing challenge. Additionally, there are ethical questions around responsible handling of data and communication to patients, in particular with direct-to-consumer test kits.
Epigenetic Sequencing
Epigenetics is an emerging field with important implications in medicine. Epigenomic features, like DNA methylation, regulate chromatin structure and influence gene expression, with documented links to diseases (Jin 2018) like cancer, autoimmune disease, and metabolic disorders, as well as processes like cellular aging (Ciccarone 2018).
NGS has been widely used to catalogue the epigenome and understand the functional consequence of these marks. Bisulfite sequencing (Barros-Silva 2018) is commonly used to profile DNA methylation, which is observed on 80% of cysteines in cysteine/glycine (CpG) regions, and has published links to cancer. Histone modifications like phosphorylation, methylation, and ubiquitination are mapped using chromatin immunoprecipitation (ChIP) followed by NGS sequencing (Sarda 2014). For studies of whole-genome chromatin accessibility the assay of transposase accessible chromatin (ATAC-seq) is faster than other methods like DNase-seq. In the ATAC-seq method (also performed in single cells), adapters for high-throughput sequencing are ligated into accessible regions using the action of a hyperactive Tn5 transposase, and mapped by sequencing (Buenrostro 2015).

sc-ATAC-Seq workflow. Single cells or isolated nuclei are tagged at regions of open chromatin using hyperactive Tn5 transposase in a bulk reaction. Barcoding and amplification of tagged sites in individual droplets generate a library of fragments representative of the original open chromatin profile of each cell.
Learn more about our scATAC-Seq products »
Functional data from these efforts are available on the NIH Roadmap Epigenomics Project and Blueprint online databases. Improved protocols and computational tools will further enhance our functional understanding of the epigenome.
Genome Editing (CRISPR/Cas9)
CRISPR/Cas9 is a powerful genome editing tool that allows precise changes to the DNA of living cells and animals. These methods have seen expanded use in the research setting, with the promise of a cure for previously untreatable genetic disorders. However, the off-target activity of these methods makes them far too risky for human therapeutics. NGS methods are widely used to detect off-target effects in vitro or in vivo (Vakulskas 2019; Zhang 2015). In vitro, digested genome sequencing methods detect double-strand breaks in cell-free genomic DNA, while in vivo methods identify double-strand breaks and associated rearrangements in different cell types under varied conditions. Data analysis remains a significant obstacle, as there is a current lack of adequate commercial solutions.
Immune Repertoire Sequencing
Cancer immunotherapies, including checkpoint inhibitors, adoptive-cell therapies, and therapeutic vaccines have demonstrated immense success in patients. However, drug resistance and limited efficacy in many patient populations necessitates a better understanding of the molecular mechanisms involved. Studies of the human immune repertoire, which is the collection of diverse T cell and B cell receptors, are used to better dissect immunotherapy responses, with the aim of designing personalized treatments to serve broader patient populations (Zhuang 2019). Immune repertoire sequencing (IRS) of DNA and RNA is favored over proteomics methods for large-scale analysis of the immune repertoire and is combined with other technologies to identify new vaccine targets. Wide application of IRS in basic science and clinical oncology hinges on advancements in existing bioinformatics tools and protocols.
Conclusion
The future of genomics belongs to rapidly evolving third-generation sequencing methods (long-read, direct-read, and single-cell) and new technologies around in situ sequencing and spatialomics. As new technologies emerge at an unprecedented rate, so do questions and challenges around data storage, quality control, regulatory guidelines, and the ethics of handling such data. Further, reduced costs and better bioinformatics approaches will help close the translation gap, and provide physicians with genomics tools for delivering timely, personalized care.
Resources
Exciting ATAC-Seq Applications
ATAC-Seq enables researchers to map open chromatin and understand the machinery behind gene regulation in exquisite detail at the single-cell level. What new applications are there for ATAC-Seq?
Dr. Jason Buenrostro of the Broad Institute, an inventor of ATAC-Seq and Bio-Rad collaborator, discusses this new exciting single-cell application.
References
Barros-Silva D, Marques CJ, Henrique R, Jerónimo C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes (Basel). 2018 Aug; 9(9):429. PMCID: PMC6162482.
Berglund E, Maaskola J, Schultz N, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018; 9(1):2419. PMCID: PMC6010471.
Bik EM. The Hoops, Hopes, and Hypes of Human Microbiome Research. Yale J Biol Med. 2016 Sep; 89(3):363-373. PMCID: PMC5045145.
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015 Jan; 109:21.29.1-21.29.9. PMCID: PMC4374986.
Burgess DJ. Spatial transcriptomics coming of age. Nat Rev Genet. 2019 Jun; 20(6):317. PMID: 30980030.
Cheng M, Cao L, Ning K. Microbiome Big-Data Mining and Applications Using Single-Cell Technologies and Metagenomics Approaches Toward Precision Medicine. Front Genet. 2019 Oct; 10:972. PMCID: PMC6794611.
Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev. 2018 Sep; 174:3-17. PMID: 29268958.
Fernandez-Marmiesse A, Gouveia S, Couce ML. NGS Technologies as a Turning Point in Rare Disease Research , Diagnosis and Treatment. Curr Med Chem. 2018 Jan; 25(3):404-432. PMCID: PMC5815091.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016; 17(6):333-51. PMID: 27184599.
Human Cell Atlas: https://www.humancellatlas.org/.
Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018; 50(8):96. PMCID: PMC6082860.
Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5(1):1–8. PMCID: PMC6147084.
Liu Z, Zhu L, Roberts R, Tong W.Toward Clinical Implementation of Next-Generation Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends Genet. 2019 Nov; 35(11):852-867. PMID: 31623871.
Mantere T, Kersten S, Hoischen A. Long-Read Sequencing Emerging in Medical Genetics. Front Genet. 2019; 10:426. PMCID: PMC6514244.
Pereira M, Malta F, Freire M, and Couto P. Application of next-generation sequencing in the era of precision medicine. In: Marchi F, Cirillo P, Mateo EC (Eds.) Applications of RNA-Seq and Omics Strategies: From Microorganisms to Human Health. InTechOpen, London, England, UK; 2017: 293–318. doi: 10.5772/intechopen.69337.
Regan JF, Kamitaki N, Legler T, et al. A rapid molecular approach for chromosomal phasing. PLoS One. 2015; 10(3). PMCID: PMC4349636.
Sarda S, Hannenhalli S. Next-generation sequencing and epigenomics research: a hammer in search of nails. Genomics Inform. 2014 Mar; 12(1):2-11. PMCID: PMC3990762.
Shinde P, Mohan L, Kumar A, et al. Current Trends of Microfluidic Single-Cell Technologies. Int J Mol Sci. 2018; 19(10):3143. doi: 10.3390/ijms19103143.
Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019; 20(11):631-656. PMID: 31341269.
Vakulskas CA, Behlke MA. Evaluation and Reduction of CRISPR Off-Target Cleavage Events. Nucleic Acid Ther. 2019 Aug; 29(4):167-174. PMCID: PMC6686686.
Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids. 2015 Nov ; 4(11):e264. PMCID: PMC4877446.
Zhuang Y, Zhang C, Wu Q, Zhang J, Ye Z, Qian Q. Application of immune repertoire sequencing in cancer immunotherapy. Int Immunopharmacol. 2019 Sep; 74:105688. PMID: 31276974.