

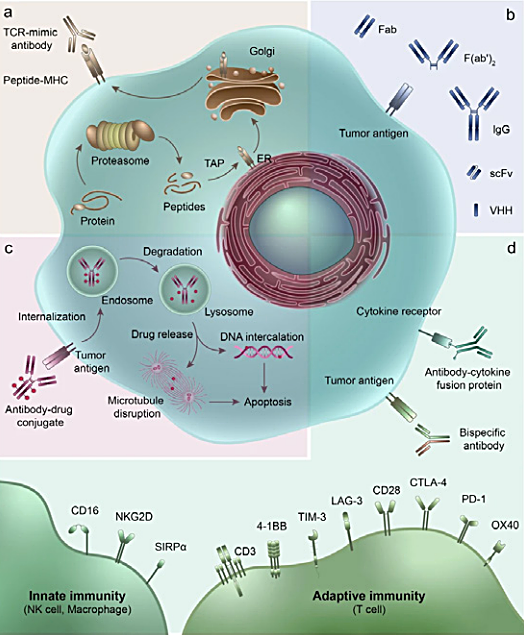
Fig. 1. Therapeutic antibodies and their derivatives. A, TCR-mimic antibody; B, IgG antibody and antibody fragments; C, ADC and mechanism of action; D, multifunctional antibodies, such as bispecific antibodies and antibody-cytokine fusion protein. Image taken from Jin et al. 2022 under a CC BY 4.0 license.
Therapeutic antibodies have transformed the precision medicine landscape due to their excellent specificity, high affinity, long in vivo half-life, and potency. These features have rendered these drugs essential for treating a wide range of debilitating diseases, including cancer, autoimmune disorders, and infectious diseases. Developing and bringing therapeutic antibodies to the market entails a complex process that involves many stages, from initial target identification to manufacturing. One of the most critical aspects of this process is ensuring the safety of therapeutic antibodies, as any adverse effects could compromise a therapeutic's success. Therefore, biopharmaceutical companies and regulatory agencies responsible for overseeing the development and approval of these drugs invest a significant amount of resources to ensure their efficacy and safety throughout all stages of development.
Therapeutic antibodies, such as monoclonal antibodies (mAbs), have emerged as a revolutionary class of biotherapeutics, which offer significant benefits over traditional small molecule drugs. Different classes and formats of therapeutic antibodies with varied mechanisms of action, including mAbs, antibody-drug conjugates (ADCs), and bispecific antibodies, are currently being employed to address unmet medical needs (Jin et al. 2022) (Figure 1). Developing and bringing these drugs to the market requires a multifaceted process involving rigorous interdisciplinary evaluations of key parameters influencing therapeutic antibody development (Figure 2). However, this is not without its challenges, and successfully demonstrating both drug efficacy and safety is critical for regulatory approval and market authorization (Sifniotis et al. 2019).

Fig. 2. Parameters for consideration during the therapeutic antibody development process. PD, pharmacodynamic; PK, pharmacokinetic.
Therapeutic Antibody Development Process Overview
Target Identification and Validation
Therapeutic antibody development process begins with identifying and validating a potential target that may be involved in a disease. In choosing a target, the probability of it being druggable must also be considered; that is, a drug must be able to bind the target and affect its function in a desirably. Therefore, a target should be accessible to the putative therapeutic antibodies and the interaction should produce a measurable biological effect — aiming to primarily establish efficacy rather than safety, which becomes the main focus at later stages of development. Target screening can be done through biophysical cell- and tissue-based evaluations using a variety of techniques including flow cytometry, genetic engineering, and in vivo assays.
Therapeutic Antibody Discovery and Optimization
Once a suitable target has been identified, the next step is antibody lead discovery and optimization, where researchers use a variety of in vitro and in vivo techniques to generate and optimize antibodies that can bind to the target with high specificity and affinity (Wang et al. 2021). Additionally, it is important to assess the stability and developability properties of lead candidates prior to proceeding to more intensive animal studies in preclinical development. These in vitro assays, such as thermal stability or binding kinetics, ascertain the top candidates for further development.
Preclinical Development
At this stage, complimentary in vitro and in vivo testing is then performed to evaluate the safety in addition to the efficacy of a lead antibody. Assessing a candidate's immunogenic, pharmacokinetic (PK), and pharmacodynamic (PD) properties is critical. Often ligand-binding assays (LBAs) are used to understand the drug's efficacy and potential toxicity, and for determining optimal dosing regimens. Anti-drug antibody (ADA) assays are also performed to detect the presence of antibodies that the immune system produces in response to the drug. These assays, which should be designed with regulatory requirements in mind (e.g., ICH M10 Bioanalytical Method Validation and Study Sample Analysis), establish the safety parameters that will guide the therapeutic throughout further development.
The quality of detection reagents used in LBAs is critical to the success of this phase of antibody development. Fully human anti-idiotypic (anti-ID) antibodies are ideal for these types of assays because they provide greater specificity than traditional antibodies (Harth et al. 2018). They also ensure higher batch-to-batch consistency and enable the development of robust assays that can be used throughout the development process, avoiding the need for continual assay validation.
Employing recombinant antibody libraries and phage display technologies for the generation and production of anti-idiotypic antibodies offers several advantages over conventional antibody generation methods (Tornetta et al. 2007). Producing anti-ID antibodies in E. coli eliminates the need for animal use throughout this process. The complete humanness of the anti-ID antibodies and availability of human isotypes renders this process useful for the development of positive controls and calibrators in human immune response assays in preclinical and clinical settings. In vitro development also allows for greater flexibility in terms of antibody selection approaches, leading to excellent antibody specificities (Harth et al. 2018). Additionally, this method allows for secure, consistent, and long-term supply of anti-ID antibodies, as backups of bacterial antibody clones and their plasmid DNA can be easily generated. Finally, with a production timeline of as little as 8 weeks, this process is considerably faster than animal-based systems, which may take up to 8 months to deliver detection reagents.
Clinical Development
Once the lead candidate has passed preclinical testing, a series of clinical trials take place to establish the safety and efficacy of the antibody in humans. Clinical trials are divided into three phases, starting with phase I trials, where the safety and tolerability of the antibody are evaluated in a small group of healthy volunteers. Phase II trials involve a larger group of patients with the disease being treated, and the efficacy of the antibody is evaluated. Phase III trials involve even larger groups of patients and are designed to confirm the safety and efficacy of the antibody.
There are several challenges associated with transitioning from lab-scale processing to process-scale drug production for large clinical trials. While upstream processing advancements have improved the production yield of target antibodies, they can also negatively impact product quality, impurity composition, and concentration upon harvest, which significantly affects downstream processing (Gronemeyer et al. 2014). For example, monoclonal antibody (mAb) overproduction is associated with an increased presence of antibody aggregates due to excess cross-bonding and the denaturing of drug molecules during typical cell culture conditions (Joshi et al. 2013).
Therefore, purification processing plays an essential role in ensuring product purity and safety during production and often involves several steps, including capture, intermediate, and polishing column chromatography, ultrafiltration, and diafiltration. The resin selected for each step must be compatible with the specific purification challenges associated with that stage of the process. The nature and downstream sequence of the purification steps following protein capture depend on the nature of the protein of interest itself and the types of impurities that need to be removed prior to product isolation, such as host cell DNA (hcDNA) and proteins (HCPs), endotoxins, viruses, and protein aggregates (Tuameh et al. 2022).
Manufacturing
Finally, if the clinical trials are successful, the antibody will be manufactured commercially using cell culture systems. The final product is purified and formulated to ensure stability and efficacy before market distribution. During this stage, antibody stability testing is conducted to ensure that antibodies remain stable during production, storage, and transit, as loss of stability can affect antibody efficacy and safety.
Innovative Tools Used to Assess Product Purity and Safety Throughout Therapeutic Antibody Development
As the diversity and complexity of therapeutic antibody products continue to grow, so do the challenges facing downstream processing, such as increased production costs due to the process complexity and increased requirements of processing resources (e.g., in column packing). Manufactured and packed with the appropriate resin under good manufacturing practice (GMP) conditions, ready-to-use prepacked process scale chromatography columns can help optimize downstream processes by eliminating the need to manually pack columns — a time-consuming and specialized activity — thus reducing variability in packing conditions and quality, leading to increased process efficiency and reproducibility. Prepacked columns can also reduce the risk of cross-contamination between batches when used as a part of a single-use continuous process system. They also provide greater flexibility in terms of sample size and allow for easier scale-up to larger volumes (see related article).
Additionally, mixed-mode chromatography has emerged as a robust tool for streamlined antibody purification — harnessing mixed-mode resins capable of at least two modes of interaction, such as hydrophobic interaction and ion exchange, can selectively remove a broad range of impurities in a single step (see related article). By designing a production workflow with carefully selected multi-mode resin prepacked columns, antibody yields can be optimized, reducing process times and increasing antibody purity.
During the drug development process, there has also been a push to rapidly collect more data on multiple parameters at once, without compromising process efficiency or data quality. As a result, over the past few years, the biopharmaceutical industry has increasingly been utilizing automated high-throughput (HT) screening tools, to support product development. HT screening involves the rapid and parallel analysis of multiple samples that have been subjected to a particular set of conditions, using a variety of analytical tools and assays, such as high-throughput flow cytometry or multiplex assays. HT screening starts with predictive modeling and spans all areas of the drug development process, from determining the ideal expression systems and growth conditions to selecting purification methods, formulation conditions, and beyond. Such tools can save researchers significant time in procuring and analyzing data, thus improving overall process development efficiency.
Monitoring product purity throughout development is essential, particularly as the purification process is scaled up. Researchers can develop safe and effective therapeutic antibodies suitable for market authorization using a range of accurate, reliable, and reproducible assays. Trace amounts of residual hcDNA may, for example, be carried over into the final product, potentially introducing the risk of immunogenicity or oncogenesis. Because of this, regulatory agencies such as the U.S. Food and Drug Administration (FDA) require therapeutic products to contain no more than 10 ng of hcDNA per dose (Yang 2013). Therefore, accurate quantification of hcDNA is essential to produce a safe and appropriate therapeutic dose. Traditional methods for hcDNA monitoring include quantitative PCR (qPCR), which is characterized by several drawbacks, such as amplification bias and nonspecific signal. Furthermore, qPCR requires sample extraction, which increases hands-on time and time to results.
In contrast, Droplet Digital™ PCR (ddPCR™) technology counts nucleic acid molecules encapsulated in thousands of individual droplets (reactions) in a single low-volume sample and does not rely on perfect amplification conditions. Because of its high inhibitor tolerance, ddPCR technology is extraction free and provides an absolute count of target DNA copies per input sample. The precision and accuracy offered by ddPCR technology also allow for significantly less variation compared to qPCR making this technique ideal for measurements of trace amounts of target DNA. This technology can also be used throughout development and manufacturing to detect viruses, Mycoplasma, and other cell culture contaminants that may impact product safety.
Conclusion
Ensuring the safety and efficacy of therapeutic antibodies is of utmost importance for their success, with bioanalytical, quality control, and purification processes playing a critical role from early-stage research and development to final production. Investment in reliable, established tools with proven success lessens risks of unexpected regulatory hurdles in addition to providing quality data.
This approach helps minimize safety risks, product recalls, and costly delays in regulatory approval. By adhering to strict guidelines and using the most advanced and innovative tools and methods, companies can ensure the highest standards of safety and efficacy for therapeutic antibodies.
References
Gronemeyer P et al. (2014). Trends in upstream and downstream process development for antibody manufacturing. Bioengineering 1, 188–212.
Harth S et al. (2018). Generation by phage display and characterization of drug-target complex-specific antibodies for pharmacokinetic analysis of biotherapeutics. mAbs 11, 178–190.
Jin S et al. (2022). Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct Target Ther 7, 39.
Joshi V et al. (2013). Aggregation of monoclonal antibody products: formation and removal. Biopharm International 26, 40–45.
Mould DR and Meibohm B (2016). Drug development of therapeutic monoclonal antibodies. BioDrugs 30, 275–293.
Sifniotis V et al. (2019). Current advancements in addressing key challenges of therapeutic antibody design, manufacture, and formulation. Antibodies (Basel) 8, 36.
Tornetta M et al. (2007). Isolation of human anti-idiotypic antibodies by phage display for clinical immune response assays. J Immunol Methods 328, 34–44.
Tuameh A et al. (2023). Methods for addressing host cell protein impurities in biopharmaceutical product development. Biotechnology Journal 18, e2200115.
Wang B et al. (2021). Optimization of therapeutic antibodies. Antib Ther 4, 45–54.
Yang H (2013). Establishing acceptable limits of residual DNA. PDA J Pharm Sci Technol 67, 155–163.
Content You'll Also Find Interesting
Recommended Features
-
Image

Detecting Contaminants During Therapeutic Antibody Production
Learn how new technologies reduce contamination risks in monoclonal antibody therapeutics manufacturing. Find tips for optimizing impurities detection.
-
Image
Overcoming Monoclonal Antibody Purification Challenges
Learn critical success factors for monoclonal antibody purification and strategies you can leverage to accelerate production.
-
Image
Advancing Cell Line Development to Streamline Biopharmaceutical Production
Explore four areas that advance cell line development for biologics including cell line selection, genetic clonality and stability, and host cell engineering.
Subscribe
Get Topic-Specific Insights
Sign up to stay informed on the latest insights and trends in your field. Receive biweekly articles, news, and more.