
Recombinant tagged purification is a widely used method for purifying proteins of interest based on well-developed recombinant DNA and protein expression technologies. Theoretically, all proteins can be purified using this method regardless of the solubility of the proteins produced in expression host cells. This section provides an overview of tagged recombinant protein affinity purification and discusses specific tagged purifications, such as polyhistidine (His) (Profinity™ IMAC resin), glutathione-S-transferase (GST), (Profinia™ GST resin), and Profinity eXact™ resin. General considerations and trouble shooting tips are discussed for each of these methods. Learn about various methods for protein purification by affinity chromatography using media based on protein A/G and other affinities or activated supports.
Related Topics: Affinity Chromatography, Protein A/G Affinity, and Activated Supports.
Page Contents
Understanding cellular processes requires knowledge of the structure, function, posttranslational modifications, and interactions of proteins. Until recently, progress in understanding proteins had been slowed by the difficulty in purifying proteins. The advent of easy-to-use and high-throughput methods for protein purification should accelerate proteomics research. Useful applications of affinity-tag purification schemes include:
- Developing and producing proteins for therapeutic applications
The human augmenter of liver regeneration (hALR), a hepatotrophic protein that can stimulate hepatic cells to grow regardless of genus, has been expressed and purified from Escherichia coli (Sheng et al. 2007). The expression of hALR enables further study of its biological function, and also suggests that recombinant hALR could be developed for repair of hepatic damage. - Developing vaccines
Purification of a spike glycoprotein tagged with histidine, one of the major structural proteins of SARS-associated coronavirus (CoV), allowed production of a large amount of the protein, which maintained antigenicity and immunogenicity and induced strong IgG responses in mice (Zhao et al. 2005).
Similarly, serine proteinase inhibitor (serpin) from Schistosoma japonicum (Sj serpin) was tagged with histidine, expressed in E. coli, and purified by affinity chromatography (Yan et al. 2005). Mice immunized with the purified protein produced high levels of specific antibodies and developed moderate protection against infection by S. japonicum. - Generating sufficient protein for crystallization and structural analysis
Studies of Mycoplasma pneumoniae aim to find novel protein structures that may have similar functions across species (Chen et al. 2004; see BioRadiations 117). - Identifying protein binding partners to elucidate functional pathways
Hardwidge et al. (2006) created GST-fusions with virulence proteins from enteropathogenic E. coli (EPEC), an enteric human pathogen responsible for much worldwide morbidity and mortality. These fusion proteins were expressed in Saccharomyces cerevisiae, and the yeast proteins that interact with the proteins were isolated by affinity purifying against the GST tag. These complexes were subjected to isotope-coded affinity tagging combined with electrospray ionization-tandem mass spectrometry, and the peptide sequences were searched against a database, which provided a list of proteins that bound specifically to each EPEC virulence protein. - Determining cellular localization, identifying posttranslational modifications, and biochemically characterizing proteins
Tristetraprolin (TTP), an anti-inflammatory protein that destabilizes mRNA, has not been adequately characterized, due to the difficulties in protein purification. Purification of a fusion protein expressed in E. coli (Cao 2004) allowed generation of antibodies that could be used for cellular localization of the protein. Additional studies measured the binding affinity of TTP under different conditions, which suggested that phosphorylation and other posttranslational modifications reduce TTP's mRNA binding affinity by half. - Characterizing protein properties via site-directed mutagenesis
Site-directed mutagenesis was used to target the putative NADP binding site of maize photosynthetic NADP-malic enzyme (Detarsio et al. 2003), which catalyzes the oxidative decarboxylation of L-malate to yield pyruvate, CO2, and NADPH. In maize and other C4 plants, this enzyme is involved in a CO2-concentrating mechanism that increases photosynthetic yield. Following expression in E. coli and purification of the recombinant protein, the participation of mutated residues in substrate binding and the catalytic reaction was inferred by kinetics and by circular dichroism and fluorescence spectra.
| Advantages | Disdvantages | |
|
|
The two most commonly used tags are the polyhistidine tag generally consisting of six 10 histidine residues and GST.
Proteins tagged with histidine bind strongly with metal ions, such as Ni2+, Co2+, Cu2+, and Zn2+. They and are purified by binding to a metal ion immobilized on a support resin by the IMAC method (immobilized metal ion affinity chromatography). Either end of the recombinant protein can be tagged with histidine. Optimal placement is empirically determined and can vary from protein to protein.
For GST tags, the purification principle is based on the binding of GST to gluthathione immobilized on the support resin. After sample impurities are washed from the resin, the bound GST-tagged protein is eluted by reduced glutathione. The GST protein has a molecular weight of 26 kD and is most often fused to the target protein at the N-terminus, though it can work well with C-terminal fusions. The GST protein is a dimer in solution and, thus, the fusion protein dimerizes as well. GST tags can be used to increase solubility of the recombinant protein.
Tag Features
| Histidine | GST | |
|
|
IMAC Troubleshooting
| Problem | Solution |
| No binding | Chelating agents such as EDTA or EGTA should not be used in purification buffers since nickel, cobalt, or copper ions are used in the purification process. Under native conditions, the histidine tag is buried within the protein and unavailable to bind. To test, bind under denaturing conditions. If the histidine tag is buried under native conditions, try fusing the histidine tag to the other end of the protein, adding another fusion tag, or adding nonsense sequence adjacent to the histidine tag. |
| Low yield | This could be due to the resin, the purification buffer/protocol, or both. It is recommended to test different IMAC resins to empirically determine the optimal resin for the protein of interest. Purification buffers that contain high concentration of imidazole may weaken the protein binding to the resin. The optimal imidazole concentration can be specific to the IMAC resin. Flow rate is critical to protein binding. Lower flow rates increase potential yield. High flow rates may not allow sufficient time for the proteins to bind and can be lost in the flow through. See Figure below. Batch binding or multiple loading can offset this problem. The cartridge or column capacity is also an important factor in protein yield. Overloading the column can reduce the protein yield. A general rule is to keep the protein load within 50–75% of the column volume. |
| Purity | Because many host proteins contain multiple tandem histidine residues, they can bind weakly to the IMAC resin and co-elute with the protein of interest. Nonspecific binding can be reduced by increasing the salt concentration (1.5 M NaCl) in the lysis and wash buffers. Add a low concentration of imidazole to the lysis and wash buffers and use an imidazole gradient to elute the protein. Host chaperone protein (HCP) is a common contaminant that is often co-purified with tagged proteins. In this case, MgCl2 and ATP can be used to remove the chaperone proteins (Rial and Ceccarelli, 2002, Protein Expression and Purification). |
GST Troubleshooting
| Problem | Solution |
| Low yield | Use lower flow rates for sample loading and elution due to the slow binding and disassociation of the GST from glutathione. Batch binding can be a good choice. |
| GST protein contamination | The cells expressing the fusion protein are not derived from a single colony. |
Bio-Rad offers a unique fusion tag purification system. The Profinity eXact fusion tag purification system is an E.coli-based expression and purification system that generates a tag-free, highly purified protein containing its native N-terminal amino acid sequence in a single step without the addition of protease.
Advantages
- Highly purified tag-free target protein
- Rapid purification and on-column cleavage
- Novel alternative to existing affinity tag and tag removal technologies
This tag system utilizes a modified form of the subtilisin protease, which is immobilized onto a chromatographic support.
The tag is the prodomain (prosignal sequence) of the subtilisin protease, a 75 amino acid sequence that is fused to the N-terminus of the target protein of interest. The mature and prodomain subtilisin protease sequences have been co-engineered to produce a highly specific, high-affinity interaction between the binding partners (KD <100 pm).
Application of the elution buffer triggers the subtilisin processing activity, which quickly and precisely cleaves the tag from the fusion protein and releases the purified protein target. The tag remains tightly bound to the resin and contains only its native amino acid sequence. The structure and activity of the native protein is maintained and the need for cleavage enzymes and purification resin for post-cleavage enzyme removal is eliminated.
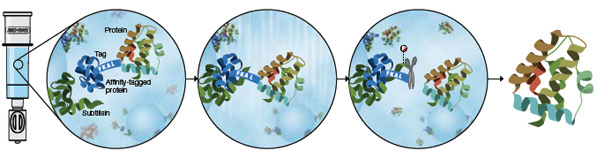
Purification and on-column tag cleavage using the Profinity eXact fusion-tag system. During sample application, immobilized subtilisin protease (ligand) recognizes and binds the affinity-tagged protein. Washing the column removes unbound contaminants. Application of fluoride-containing elution buffer triggers subtilisin to quickly and precisely cleave the tag from the fusion protein after the FKAL cleavage recognition sequence. The tag remains tightly bound to the resin, and a highly purified protein with only its native amino acid sequence is released.
Cao H, Expression, purification, and biochemical characterization of the antiinflammatory tristetraprolin: a zinc-dependent mRNA binding protein affected by posttranslational modifications, Biochemistry 43, 13724–13738 (2004)
Chen S et al., Crystal structure of a protein associated with cell division from Mycoplasma pneumoniae (GI: 13508053): a novel fold with a conserved sequence motif, Proteins 55, 785–791 (2004)
Detarsio E et al., Maize C4 NADP-malic enzyme. Expression in Escherichia coli and characterization of site-directed mutants at the putative nucleotide-binding sites, J Biol Chem 278, 13757–13764 (2003)
Hardwidge PR et al., Proteomic analysis of the binding partners to enteropathogenic Escherichia coli virulence proteins expressed in Saccharomyces cerevisiae, Proteomics 6, 2174–2179 (2006)
Sheng J et al., Cloning and expression of the human augmenter of liver regeneration at low temperature in Escherichia coli, J Biochem Biophys Methods 70, 465–470 (2007)
Yan Y et al., Characterization of a novel vaccine candidate and serine proteinase inhibitor from Schistosoma japonicum (Sj serpin), Vet Parasitol 131, 53–60 (2005)
Zhao JC et al., Prokaryotic expression, refolding, and purification of fragment 450–650 of the spike protein of SARS-coronavirus, Protein Expr Purif 39, 169–174 (2005)