

Targeted genome editing has become a powerful genetic tool for studying gene function, for modifying genomes to correct defective genes, or to introduce new functionality. The mechanism of targeted genome editing involves sequence-specific breaks in the target DNA, with edits incorporated during repair. Genome editing is used in a variety of applications, including gene knockout, repression of gene activation, drug discovery screens, and imaging genomic loci.
Different systems are used for targeted editing including CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9). This system’s ability to create targeting constructs in days rather than weeks or months, its low cost relative to other targeted editing systems, and its adaptability have opened up a wide range of possibilities for researchers, biotechnology companies, and drug discovery.
Page Contents
All genome editing systems rely on sequence-specific cleavage by endonucleases. There are two types of mechanisms for sequence-specific targeting: DNA guided and RNA guided. The most commonly used DNA-guided systems are meganucleases (MNs), zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs). The most common RNA-guided system is the CRISPR/Cas9 system (Cas9 is an endonuclease).
Meganucleases (MNs)
MNs, found in bacteria, are nucleases that have long recognition sequences, making them highly specific. The longer the recognition sequence, the lower the probability that there will be off-target events. MNs can be engineered to recognize specific sequences, but it is costly and time-consuming.
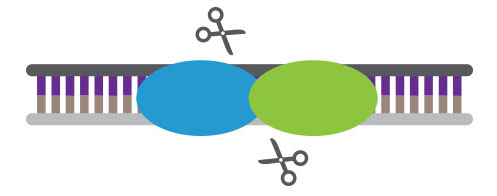
Meganucleases are highly specific and easy to deliver to cells but difficult to redesign for new targets.
Zinc Finger Nucleases (ZFNs)
ZFNs are created by fusing a zinc finger DNA-binding domain, a small structural motif stabilized by coordinated metal ions, to the DNA-cleavage domain of the nonspecific FokI endonuclease. Zinc finger motifs are designed to bind to specific DNA sequences, which are then cleaved by the nuclease active site.
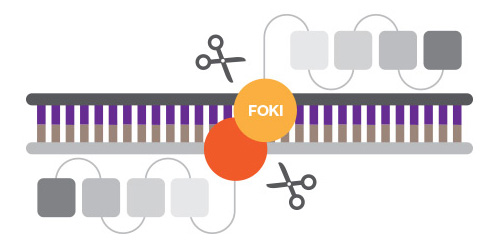
Zinc finger nucleases have been more difficult to design than anticipated given their modular nature.
Transcription Activator-Like Effector Nucleases (TALENs)
TALENs are chimeric proteins that function like ZFNs, but the sequence-specific DNA-binding domain is a TAL (transcription activator-like) effector, and DNA cleavage occurs via FokI or other nucleases' active site. TALENs are easier to engineer than ZFNs as the sequence specificity is determined by only two amino acids, while design and synthesis of zinc finger motifs that recognize nucleotide sequence with high specificity requires more protein engineering.
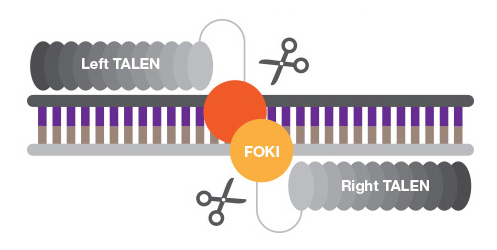
TALENs are truly modular and not limited by sequence requirements, but their large size makes cell delivery challenging.
CRISPR/Cas9
CRISPR/Cas9 targeted genome editing techniques have been developed from systems that evolved in prokaryotes to help provide immunity from viruses such as bacteriophages and other foreign DNA, such as plasmids (Horvath and Barrangou, 2010). There are several variants of the CRISPR/Cas9 system, with different mechanisms, described below.
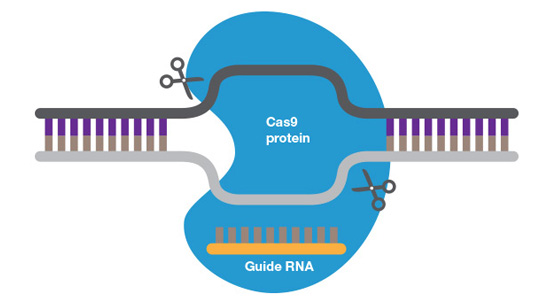
CRISPR/Cas9 complexed with genomic DNA.
Comparison of Gene Editing Techniques
| Meganucleases | Zinc Finger Nucleases | TALENs | CRISPR/Cas9 | |
| Target site length | 14–40 bp | 18–36 bp per ZFN pair | 28–40 bp per TALEN pair | 19–22 bp |
| Specificity | Some mismatches are tolerated | Some mismatches are tolerated | Some mismatches are tolerated | Tolerant even of several consecutive mismatches |
| Off-target editing | Low | Low to moderate | Low | Common without additional homologous sequence* |
| Targeting constraints | Reengineering for new specificities is challenging | G-rich regions are challenging | 5' T for each TALEN is required | PAM** is required |
| Ease of design | Extremely difficult | Difficult | Moderate | Easy |
| Multiplexing | Challenging | Challenging | Challenging | Easy |
| Ease of in vivo delivery | Small size allows use of many viral vectors | Small size allows use of many viral vectors | Large size limits viral vectors; repetitive sequences lead to unwanted recombination | Plasmid or viral vectors |
| Ease of ex vivo delivery | Relatively easy | Relatively easy | Relatively easy | Relatively easy |
*Addition of sequence to guide RNA that is homologous to upstream and downstream sequences
** Protospacer Adjacent Motif; a short sequence downstream of the target DNA sequence
Structure of CRISPR/Cas9 and Sequence Requirements
The CRISPR/Cas9 system is composed of a single guide RNA (sgRNA) and Cas9, a double-strand nuclease. sgRNA is a complex of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA or trRNA). crRNA, usually about 20 bp long, contains a sequence complementary to the target region of the genome and may contain a non-homologous sequence to create a specific edit (see below). The tracrRNA hybridizes to the crRNA, creating an active complex that on binding to Cas9 converts it from an inactive into an active DNA-binding conformation. In addition to target sequence complementary to the crRNA, the protospacer adjacent motif (PAM), a specific sequence of 3 to 5 nucleotides downstream of the target sequence, is required for Cas9 binding and DNA cleavage. Different Cas9 nucleases have different PAM sequences. Where there is a target sequence and a PAM sequence in proximity, the CRISPR/Cas9 complex will bind and cleave the DNA resulting in blunt or near blunt ends.
Reduction of Off-target Events using Cas9 Nickases
Since the length of the sequence homology for CRISPR/Cas9 is only ~ 20 bp, there is a high probability of one or more off-target events due to the same sequence being found in multiple regions of a genome. To reduce these off-target events, some Cas9 endonucleases have been engineered to have a single-strand nickase instead of DSB activity. Two CRISPR/Cas9 constructs can be generated with the Cas9 nickases targeting opposite strands. Since there are now two different sequence homologies (one for each Cas9 that must be adjacent to the targeted area), this reduces the probability of off-target events.
DNA Repair Mechanisms after Cleavage by Cas9
After DNA cleavage, a double strand break (DSB) is repaired by one of two cellular mechanisms, non-homologous end joining (NHEJ) or homology directed repair (HDR).
NHEJ Repair
NHEJ is more efficient than HDR, but has much lower fidelity and, therefore, is much more error prone, resulting in insertions or deletions (indels). NHEJ requires less sequence homology than HDR, does not require a repair template, entails less DNA synthesis, and is faster. Additionally, unlike HDR, NHEJ occurs throughout the cell cycle. After accurate repair, the sequence can be re-cleaved and repaired again. This leads to an increasing proportion of cells with indels that are heterogeneous. This can provide a population of cells with mutations that cause loss of gene function, have residual function, or sometimes gain of function. Due to the introduction of errors by NHEJ, it may not be necessary to have any non-homologous target sequence in a construct to generate knockouts.
HDR Repair
HDR requires longer sequence similarity than NHEJ, which requires alignment of only a few complementary bases for the ligation of the two ends. If the sequence is completely homologous, HDR has a very low error rate. Since an accurate repair is a target for re-cleaving by CRISPR/Cas9 with the longer sequences used for HDR, mismatched bases can be introduced into a CRISPR/Cas9 construct to inhibit further editing. For instance, mutations can be introduced into the PAM sequence, preventing binding of Cas9.
HDR is most efficient if the DSB and modification site are within 10 nucleotides. If the aim of genome editing is to make a specific change in a gene such as a point mutation, then the design of the CRISPR/Cas9 construct will need to promote HDR over NHEJ.
To increase the level of repair by HDR, the repair template can be engineered to have additional homologous sequence both upstream (left homology arm) and downstream (right homology arm) of the targeted area. HDR is less efficient than NHEJ, so there will be a reduced yield of repair. Additionally, there will still be a low level of NHEJ activity, and, hence, the possibility (albeit very low) of an error.
Horvath P and Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–70. PMID: 20056882
More Information
Quinlan A (2017). Erasing disease: How gene editing is changing genetic therapies., accessed April 4, 2017
Quinlan A (2016). CRISPR: Changing the pace of BioPharma R&D., accessed April 4, 2017
Quinlan A (2016). From megaTALs to CRISPR: The many ways to edit a gene., accessed April 4, 2017