
The electrophoretic separation of proteins can be achieved by various methods, matrices, and buffer systems. By choosing suitable separation matrices and corresponding buffer systems, one can optimize the separation. This page provides an overview of the different electrophoretic methods that can be used.
Related Topics: Protein Electrophoresis Reagent Selection and Preparation, and Protein Detection and Analysis.
Page Contents
Acrylamide gels serve as a size-selective sieve during separation. As proteins move through a gel in response to an electric field, the smaller molecules travel more rapidly than larger proteins (see figure below).
In most PAGE applications, the gel is mounted between two buffer chambers, and the only electrical path is through the gel. Usually, the gel has a vertical orientation, and the gel is cast with a comb that generates wells in which the samples are applied (see below). Applying an electrical field across the buffer chambers forces the migration of protein into and through the gel.
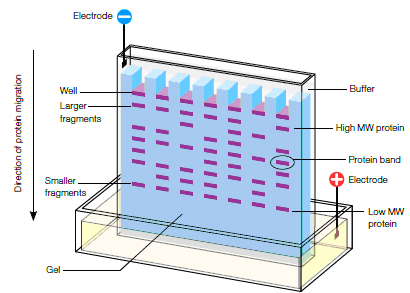
Schematic of electrophoretic protein separation in a polyacrylamide gel.
Two types of buffer systems can be used:
- Continuous buffer systems — use the same buffer (at constant pH) in the gel, sample, and electrode reservoirs. Samples are loaded into wells, and proteins that are closer to the gel enter first. This provides a uniform separation matrix, but yields fuzzy and unresolved protein bands. Continuous systems are rarely used for protein electrophoresis but commonly used for nucleic acid analysis
- Discontinuous buffer systems — use a gel separated into two sections (a large pore stacking gel on top of a small pore resolving gel, see figure below) and different buffers in the gels and electrode solutions. Proteins migrate quickly through the large pore stacking gel and then are slowed as they enter the small pore resolving gel. The proteins stack on top of one another to form a tight band, which helps improve resolution. Discontinuous buffer systems provide higher resolution than continuous systems, and varying the buffers used in the sample, gel, and electrode chambers creates a variety of discontinuous buffer systems that can be used for a variety of applications
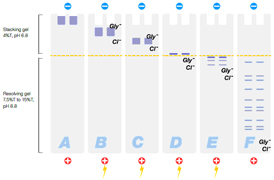
|
A: Denatured sample proteins are loaded into the wells |
Figure: Migration of proteins and buffer ions in a denaturing discontinuous PAGE system.

The original discontinuous gel system was developed by Ornstein and Davis (1964) for the separation of serum proteins in a manner that preserved native protein conformation, subunit interactions, and biological activity. Proteins are prepared in nonreducing, nondenaturing sample buffer, and electrophoresis is performed in the absence of denaturing and reducing agents. The native charge is preserved and proteins can migrate towards either electrode, but yields unpredictable separation patterns that are not suitable for molecular weight determination. Nevertheless, native PAGE does allow for separation of proteins in their active state and can resolve proteins of the same molecular weight.
To overcome the limitations of native PAGE systems, Laemmli (1970) incorporated the detergent sodium dodecyl sulfate (SDS) into a discontinuous denaturing buffer system, creating what has become the most popular form of protein electrophoresis, SDS-PAGE.
When proteins are separated in the presence of SDS and denaturing agents, they become fully denatured and dissociate from each other (see figure below). In addition, SDS binds noncovalently to proteins in a manner that imparts:
- An overall negative charge on the proteins. Since SDS is negatively charged, it masks the intrinsic charge of the protein it binds
- A similar charge-to-mass ratio for all proteins in a mixture, since SDS binds at a consistent rate of 1.4 g SDS per 1g protein SDS (a stoichiometry of about one SDS molecule per two amino acids)
- A long, rod-like conformation on the proteins instead of a complex tertiary shape
As a result, the rate at which an SDS-coated protein migrates in a gel depends primarily on its size, enabling molecular weight determination.
The original Laemmli system incorporated SDS in the gels and buffers, but SDS is not required in the gel. SDS in the sample buffer is sufficient to saturate proteins, and the SDS in the cathode buffer maintains the SDS saturation during electrophoresis. Precast gels (manufactured gels such as Bio-Rad's Ready Gel®, Mini-PROTEAN®, and Criterion™ Precast Gels) do not include SDS and can be used for either native or SDS-PAGE applications. A range of gel and buffer combinations can be used for native and SDS-PAGE, each with its own advantages (see Electrophoresis Reagent Selection & Preparation for more details).
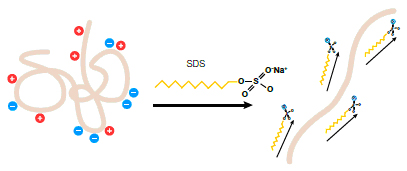
Effect of SDS on the conformation and charge of a protein.
Blue Native PAGE (BN-PAGE)
BN-PAGE is used to separate and characterize large protein complexes in their native and active forms. Originally described by Schagger and von Jagow (1987), this technique relies on the solubilization of protein complexes with mild, neutral detergents and the binding of negatively charged Coomassie (Brilliant) Blue G-250, stain to their surfaces. This imparts a high charge-to-mass ratio that allows the protein complexes to migrate to the anode as they do in SDS-PAGE. Coomassie Blue does not, however, denature and dissociate protein complexes the way SDS does.
Zymogram PAGE
Zymogram PAGE is used to detect and characterize collagenases and other proteases within the gel. Gels are cast with gelatin or casein, which acts as a substrate for the enzymes that are separated in the gel under nonreducing conditions. The proteins are run with denaturing SDS in order to separate by molecular weight. After renaturing the enzymes and then allowing them to break down the substrate, zymogram gels are stained with Coomassie (Brilliant) Blue R-250 stain, which stains the substrate while leaving clear areas around active proteases.
Isoelectric Focusing (IEF)
IEF combines the use of an electric field with a pH gradient to separate proteins according to their isoelectric point (pI). When a protein moves through a pH gradient, its net charge changes in response to the pH it encounters. Under the influence of an electric field, a protein in a pH gradient migrates to a position until its net charge is zero (see 2-D Electrophoresis for more details).
2-D Electrophoresis
The sequential application of different electrophoresis techniques produces a multi-dimensional separation. The most common 2-D technique (O'Farrell 1975) subjects protein samples first to denaturing IEF on a tube gel or IPG gel strip (for separation by pI), and then to SDS-PAGE for further separation by molecular weight. High-resolution 2-D methods enable separation of thousands of polypeptides in a single slab gel. The resulting spots can be visualized by gel staining, or they can be transferred to a membrane support for total protein staining or analysis with specific antibody detection. (See 2-D Electrophoresis for more details).
Laemmli UK (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685.
O'Farrell PH (1975). High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250, 4007–4021.
Ornstein L and Davis BJ (1964). Disc electrophoresis-I: Background and theory. Ann NY Acad Sci 121, 321–349.
Hames BD (1998). Gel Electrophoresis of Proteins: A Practical Approach (New York: Oxford University Press)
McLellan T (1982). Electrophoresis buffers for polyacrylamide gels at various pH. Anal Biochem 126, 94 – 99.
Schägger H and von Jagow G (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166, 368 – 379.
Vavricka SR (2009). Serum protein electrophoresis: an underused but very useful test. Digestion 79, 203 – 210.
Wheeler D et al. (2004). Discontinuous buffer systems operative at pH 2.5 – 11.0, 0°C and 25°C. Electrophoresis 25, 973–974.