
On This Page |
Liquid Chromatography Techniques | Liquid Chromatography Workflow | Liquid Chromatography Considerations | Liquid Chromatography Resources |
Chromatography is used to separate proteins, nucleic acids, or small molecules in complex mixtures. Liquid chromatography (LC) separates molecules in a liquid mobile phase using a solid stationary phase. Liquid chromatography can be used for analytical or preparative applications. Here, we restrict our discussion to column liquid chromatography techniques and considerations.
In column liquid chromatography, as the liquid mobile phase passes through the column, components in the mobile phase interact to varying degrees with the solid stationary phase, also known as the chromatography media or resin. Molecules of interest in the mobile phase are separated based on their differing physicochemical interactions with the stationary and mobile phases. These interactions can be based on various properties of the molecules to be separated:
- Separation by molecular size: Size Exclusion Chromatography (SEC)
- Separation by charge: Ion Exchange Chromatography (IEX)
- Separation by hydrophobicity: Hydrophobic Interaction Chromatography (HIC)
- Separation by specific binding interactions: Affinity Chromatography
- Separation by selective combined interactions: Multimodal or Mixed-Mode Chromatography
The compostion of the mobile phase is typically changed during a separation run so as to alter the strengths of the interactions of the compounds of interest, that is, to change the phase partitioning of each compound between the stationary and mobile phases. Each compound then elutes from the column in a particular order depending on the relative strengths of its interaction with the resin and the mobile phase.
As the mobile phase continues to flow through the column, the column effluent, or eluate, is typically collected in fractions while monitoring the concentrations of the compounds eluted from the column over time to yield an elution curve, or chromatogram. The mode of detection varies with the analyte to be detected. For protein separations by column chromatography, protein concentration can be monitored manually using a dye-based protein assay such as the Bradford assay; however, such manual monitoring is labor intensive.
More commonly, a UV detector or spectrophotometer is attached to the chromatography system to continuously monitor protein elution from the column by measuring light absorption at 280 nm (A280) by the amino acid tryptophan. The resulting chromatogram is then analyzed to quantitate proteins in the eluate. Each distinct peak represents a unique component resolved by the column, and the area under the curve corresponds to the amount of that compound eluted from the column. It is important to note that a single peak may contain more than one protein species; therefore, further analysis of the eluted fractions may be required, for example, by gel electrophoresis.
A typical chromatogram displays an initial broad peak of eluted protein that interacts weakly with the resin or, in some cases, not at all. In most cases, the column is washed with binding buffer until this first flowthrough peak (labeled “Binding Conditions” in Figure 1) has completely eluted, and the A280 reading returns to baseline.
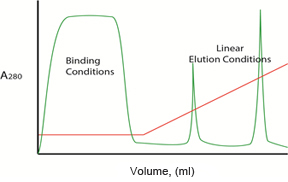
Fig. 1. Example chromatogram showing a linear gradient elution.
Proteins that interact strongly with the resin are then eluted by changing the composition of the elution buffer. The specific composition of the elution buffer depends on the physicochemical properties of the molecules to be separated and the chromatography media used, often referred to as different media chemistries. Ion exchange chromatography, for example, which relies on interactions based on the net charges of the molecules, uses elution buffers of increasing ionic strength.
The red line in Figure 1 shows a linear gradient elution, but proteins can also be eluted using stepwise isocratic elution, where buffer conditions are changed in a stepwise fashion. By altering the elution gradient, flow rate, column length, and resin particle size, the protein separation ability (or resolution) of a given resin can be changed. Chromatography methods are optimized to yield sharp, tall peaks that are well separated (Figure 2).
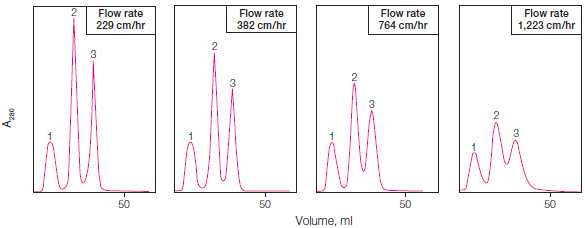
Fig. 2. Optimization of resolution by changing flow rate. Optimal peak separation is achieved at a flow
Regardless of the interactions that are being exploited, liquid column chromatography is carried out in six steps:
- Column equilibration
- Sample loading
- Washing
- Elution
- Final column washing
- Column regeneration
Column Equilibration
Most liquid chromatography protocols begin with a resin equilibration step. A buffer that is compatible with the protein of interest and the resin of choice is passed over the column. A common practice is to equilibrate the column with 5–10 column volumes (CVs) of equilibration buffer.
For example, binding of proteins to hydrophobic interaction resins is most efficient at high ionic strength. Prior to sample application, the resin is therefore equilibrated in a buffer of high ionic strength.
The properties of the protein of interest are also considered in equilibration buffer selection, as buffer factors such as ionic strength are limited by protein stability; typically, one would avoid equilibration buffer conditions that would denature the protein of interest or prevent it from interacting with the stationary phase.
Sample Loading
After equilibration, the sample is loaded onto the column. The sample is generally loaded in a buffer with the same composition as the equilibration buffer to maximize protein interaction with the stationary phase.
Sample can be loaded manually or using a sample pump. Some types of chromatography limit the volume of sample that can be loaded onto the column. Another important sample loading consideration is that most resins have a finite capacity to bind protein; overloading a column by applying too much sample can adversely affect separation.
Column Washing
Once proteins have been immobilized on the stationary phase, proteins that interact only weakly or nonspecifically with the resin are removed by washing the column with several column volumes of wash buffer. This wash buffer can have the same composition as the equilibration buffer or contain components that disrupt weak specific interactions.
For example, immobilized-metal affinity chromatography (IMAC) elutes proteins bound to the resin with a high concentration of immidazole. A common practice is to use a wash buffer that includes an intermediate concentration of immidazole to eliminate contaminating proteins that are only weakly bound to the resin.
The column is washed until no protein is detected in the eluate. When using a chromatography system with a UV detector, the column is washed until the 280 nm absorption reading returns to the baseline.
Sample Elution
After all nonspecifically and weakly interacting proteins have been washed off of the resin, proteins that interact strongly with the resin are eluted from the column by changing the composition of the buffer that is passed over the resin.
In ion exchange chromatography, proteins are eluted with high–ionic-strength buffers or with a change in pH to disrupt the electrostatic interactions that immobilized the protein of interest. Proteins bound to a hydrophobic interaction resin, conversely, are eluted by lowering the ionic strength of the buffer. In affinity chromatography, proteins are commonly eluted from the column by the introduction of a competing ligand or by cleaving the affinity tag and may also be eluted using high-salt buffers or altering pH. Other elution protocols may involve mixing solvents of varying polarity to tune the solubility of each component in the mobile phase.
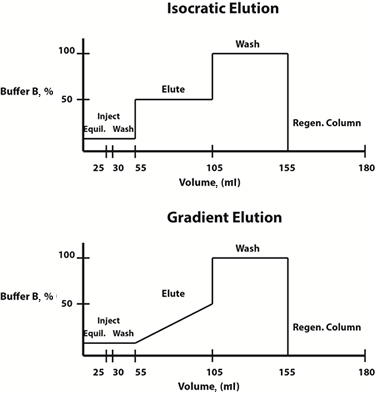
Fig. 3: Example buffer composition diagrams for isocratic (top) and gradient (bottom) elution protocols.
Elution conditions can either be changed in a linear gradient fashion or in a stepwise fashion. Often, a gradient elution protocol, in which the composition of the mobile phase changes linearly over time, is chosen to determine the elution profile and the elution buffer concentration at which the protein of interest is freed from the resin. Once this concentration has been determined, to save time, a stepwise isocratic elution protocol, in which the composition of the mobile phase is constant at each step, can be designed for future purifications.
Note: Size exclusion chromatography does not require buffer changes since it does not depend on specific interactions between the mobile phase and the stationary phase. There are no true wash and elution steps, as SEC relies solely on the fact that large molecules are retarded by porous beads, whereas small molecules pass through the resin with minimal resin interaction.
Final Column Washing
After the protein of interest has been eluted from the resin, any proteins that remain bound to the resin are eluted by increasing the strength of the elution buffer. This step permits columns to be reused for future separations.
Column Regeneration
After stripping the remaining compounds bound to the media, the column is then either saturated with equilibration buffer for subsequent reuse or filled with a storage buffer.
Four factors are important when designing protein purification workflows: resolution, yield, sample integrity, and sample purity.
Resolution refers to the separation of peaks in a chromatogram. The purpose of chromatography is to separate molecules of interest. Resolution is affected by the selective properties of the resin, by equilibration, wash, and elution buffer composition, flow rate, and by the sample volume. Compounds that elute as distinct peaks with a particular column and elution protocol may co-elute as a single peak with another chromatographic technique.
If the goal of chromatographic separation is purification of a protein of interest for downstream applications (that is, preparative chromatography), then yield, defined as the amount of the desired protein fraction recovered, is an important consideration.
Sample integrity is another key consideration for preparative chromatography. Applications such as crystallography require full-length, correctly folded protein. If activity of the protein is to be assessed in vitro, the purified protein must retain its enzymatic activity. Buffer choice, the addition of appropriate protease inhibitors, and speed are common, but not always sufficient, measures to maintain sample integrity.
Lastly, sample purity is an important consideration. In the case of co-eluting compounds, the detection of a single peak in a chromatogram does not ensure pure sample. It is therefore necessary to assess sample purity by gel electrophoresis (SDS-PAGE) or other analytic techniques.
When developing a purification workflow it is wise to consider the sample purity that is required for the intended downstream applications because sample purity, integrity, and yield often display an inverse relationship. For example, a five-column workflow may yield exquisitely pure protein, but because of the length of time required to separate the protein of interest from contaminating proteins and proteases, the protein may be completely inactive. In addition, since some of the protein of interest is lost at each column fractionation step, the total amount of protein recovered after five columns may be insufficient for the desired downstream applications.
Absolute sample purity is essential in certain applications such as antibody production for diagnostic or therapeutic applications. Some enzymatic studies, however, may require only functional purity; proteins that do not interfere with or enhance the protein of interest’s activity may be tolerated as contaminants to maximize sample integrity and yield.
Videos

This video presentation covers the basic principles of ion exchange chromatography including media choice, buffer selection, and factors that impact resolution.

This video presentation is an introduction the principles of size exclusion chromatography including sample preparation, method development, and factors affecting resolution.