
Epigenetic processes control gene expression by altering chromatin structure. Actively transcribed genes are associated with accessible chromatin regions, while transcriptionally silent genes are often in inaccessible chromatin regions. The modifications made to DNA and proteins that impact chromatin structure are referred to as epigenetic markers (or marks). Examination of histone acetylation patterns has demonstrated a high correlation between histone acetylation and active transcription, whereas histone methylation can be associated with the activation or silencing of genes depending on the amino acid modified and the number of methyl groups added.
Related Topics: Epigenetics and Chromatin Structure.
Page Contents
Histones order DNA into structural units called nucleosomes. Histones are the major protein component of chromatin and are subject to several different post-translational modifications, including methylation, acetylation, phosphorylation, ubiquitylation, and sumoylation.
The positive charge of the lysine residue on histone tails usually leads to a tight association with negatively charged DNA. Histone acetyltransferases (HATs) are a class of enzymes that instigate the formation of euchromatin from heterochromatin. HATs catalyze the transfer of an acetyl group (CH3CO) to a conserved lysine on the histone tail. Acetylation of histones typically leads to relaxed chromatin and is associated with activation of transcription. In contrast, histone deacetylases (HDACs) catalyze the removal of acetyl groups from histones, consequently leading to more tightly packaged chromatin. The recruitment of HATs and HDACs is tightly regulated by DNA binding proteins located near epigenetic target regions.

Histone Methylation. The methylation of specific amino acids on histones can lead to the formation of heterochromatin with consequent repression of transcription.
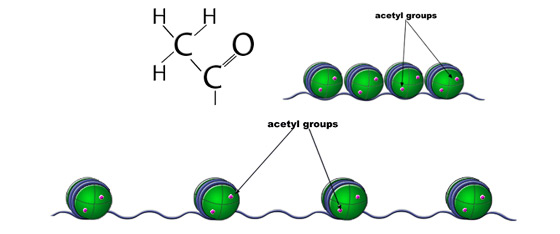
Histone Acetylation. Acetylation of histones typically leads to relaxed chromatin and is associated with activation of transcription.
Various histone modifications likely function in differing ways; acetylation at one position may have a different effect than acetylation at another position. Multiple modifications exist simultaneously and are likely working together to influence chromatin state and gene expression. The concept of multiple dynamic modifications regulating gene expression in a systematic and reproducible fashion is known as the histone code. The histone code hypothesizes that, in addition to impacting chromatin conformation, epigenetic markers significantly impact the recruitment of other proteins that modulate gene expression.
In addition to the impact of histone modifications on chromatin structure, differential gene expression is also regulated by DNA methylation. Enzymes that methylate the 5'-position of cytosine bases in DNA can lead to the formation of heterochromatin and transcriptional repression. DNA methyltransferases (DMNTs), enzymes similar to the ones that transfer methyl groups to histones, can selectively methylate cytosines in the promoter and coding region of genes, usually with the effect of blocking gene transcription. DNA methylation often leads to the compaction of chromatin via the recruitment of enzymes that methylate histones, creating an additional level of transcriptional repression. This link between DNA methylation and histone methylation represents a further, synergistic repression of gene expression in affected areas of the genome.
In addition to the 5'-methylcytosine modifications to DNA described above, enzymes can also catalyze the formation of 5'-hydroxymethylcytosines in DNA. In contrast to methylcytosine modifications, hydroxymethylcytosines are often associated with open chromatin and found in gene promoter regions (Ficz et al, 2011). Differential expression of methylcytosine and hydroxymethylcytosine DNA modifications likely play a significant role in dictating gene expression patterns throughout cell development.
Bisulfite Conversion
DNA methylation can be detected using several different approaches. A common technique for assessing DNA methylation involves the use of sodium bisulfite chemistry to differentially convert unmethylated cytosine residues to uracil, while methylated cytosines are left unmodified. Methylated cytosines can then be identified through various downstream nucleic acid analysis methods, including PCR, qPCR, and sequencing. This commonly used preparatory technique can both identify individual methylation sites and quantify the level of methylation in a particular genomic region.
High Resolution Melt Analysis
High resolution melt (HRM) analysis is the qualitative analysis of a DNA fragment's melt curve following PCR amplification. It is a simple, cost-effective, and rapid tool for studying genetic variation. HRM applications include single nucleotide polymorphism (SNP) analysis, mutation scanning, species identification, and methylation analysis. To employ HRM as an epigenetic tool, DNA is first treated with sodium bisulfite to convert unmethylated cytosine residues to uracil, followed by PCR amplification. The PCR product is then subjected to decreasing temperatures and the melt temperature and melt curve profile are obtained. Comparisons are made to known standard DNA samples to determine the relative level of methylation. The varied melt temperature from PCR products originating from bisulfite treated DNA is indicative of DNA methylation status.
Methylated DNA Immunoprecipitation
Methylated DNA immunoprecipitation (MeDIP) enables researchers to examine genome-wide changes in DNA methylation patterns. In this technique, DNA is isolated from cells and sheared using sonication. Antibodies specifically targeting methylated DNA fragments are used to isolate methylated regions. The methylated DNA fragments can be identified using high-resolution DNA microarrays or next-generation sequencing techniques. This technique allows researchers to identify global changes that occur in DNA methylation patterns across varied cells.
2. Analysis of DNA/Protein Interactions
The interaction between proteins (for example, histones) and DNA is most often studied using a technique known as chromatin immunoprecipitation (ChIP). ChIP is a preparatory method that requires the use of highly specific antibodies directed against DNA-binding proteins and can be followed by a number of nucleic acid analysis techniques, including PCR, qPCR, sequencing, and microarray hybridization. It can help determine whether certain proteins are associated with specific genomic regions and is also useful for identifying regions of the genome associated with specific histone modifications.
3. Chromatin Accessibility and Conformation Assays
Similar to limiting access to transcription factors, tightly packaged chromatin is also inaccessible to other proteins. Native chromatin states can be experimentally identified using assays that interrogate nuclease accessibility of chromatin. Nucleases are enzymes that digest nucleic acids into their monomer constituents. However, nucleases are unable to digest closed, inaccessible chromatin, leaving the DNA available for subsequent experimental examination.

Chromatin accessibility. DNA in closed heterochromatin is inaccessible to nucleases and is thus resistant to digestion. Conversely, DNA in open euchromatin is accessible to nucleases and is therefore vulnerable to nuclease mediated digestion.
EpiQ™ Chromatin Analysis Kit
The EpiQ™ chromatin analysis kit is a real-time qPCR assay for the quantitative assessment of chromatin accessibility. This assay directly measures the accessibility of a DNA region by employing a nuclease treatment followed by qPCR. In this assay, heterochromatin is inaccessible to the nuclease, rendering it protected from nuclease digestion and available for subsequent qPCR. EpiQ analysis for heterochromatin reveals a minimal shift in PCR product quantity between nuclease treated and untreated samples. In contrast, euchromatin is accessible to the nuclease, digested and unavailable for subsequent qPCR. EpiQ analysis of euchromatin shows a large quantification cycle (Cq) shift between nuclease treated and untreated samples.

The EpiQ chromatin analysis kit utilizes nuclease accessibility to discriminate open vs. closed chromatin regions. Amplification of proximal promoter regions for the epigenetically silenced HBB (reference) gene or the constitutively expressed GAPDH (target) gene was carried out in HeLa cells using the EpiQ kit and EpiQ™ chromatin SYBR® Green supermix on the CFX96™ real-time PCR detection system. A, closed chromatin regions were protected from nuclease digestion and remained intact prior to amplification, resulting in minimal quantification cycle (Cq) delays (ΔCq = 0.58) following nuclease treatment; B, open chromatin regions were susceptible to nuclease digestion and were unavailable for amplification, leading to significant Cq delays (ΔCq = 8.08) after nuclease treatment. A comparison of ΔCqs with the amplification efficiencies for each gene target factored in was used to determine the accessibility of the target gene, calculated to be >99% for GAPDH. RFU, relative fluorescence units.
Digital DNase and DNase Seq
Experimental protocols have also been developed to enable genome-wide analysis of epigenetic changes. Digital DNase and DNase Seq are two methods that employ nuclease digestion to generate short DNA strands from nuclease-accessible genomic regions. These short DNA strands are isolated and examined by next generation sequencing to provide detailed sequence information on genome stretches accessible to nuclease digestion. Researchers are employing these techniques to compare DNA-structural differences between samples on a genome-wide scale.
Chromosome Conformation Capture (3C)
Spatial changes in the nuclear localization of genomic regions is thought to influence gene expression. Chromatin looping brings sites from varying chromosome locations in close proximity. It is proposed to facilitate gene transcription by facilitating the transfer of RNA polymerase between two genomic regions. The chromosome conformation capture (3C) assay assesses the three-dimensional conformation of chromatin by examining chromatin looping. In this assay, chromatin is crosslinked, cells are lysed, and then exposed to restriction digest. Gene-specific PCR primers are used to examine looping of genomic regions hypothesized to be in close proximity in certain cellular states (reviewed in Gavrilov et al, 2009). Modifications of the 3C assays have been developed to enable additional epigenetic analyses. The circular chromosome conformation capture (4C) enables researchers to assess interactions between genetic loci in a high-throughput fashion (Ohlsson et al, 2007). Chromosome Conformation Capture Carbon Copy (5C) also provides a high-throughput analysis of genomic interactions by using next generation sequencing technologies to identify novel DNA regions in close proximity within the cell (Dostie et al, 2006).
Dostie J et al. (2006). Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 10, 1299–1309. PMID: 16954542
Ficz G et al. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473, 398–402. PMID: 21460836
Gavrilov A et al. (2009). Chromosome conformation capture (from 3C to 5C) and its ChIP-based modification. Methods Mol Biol 567, 171–188. PMID: 19588093
Ohlsson R and Göndör A (2007). The 4C technique: the 'Rosetta stone' for genome biology in 3D? Curr Opin Cell Biol 19, 321–325. PMID: 17466501
Rao S et al. (2001). Chromatin remodeling, measured by a novel real-time polymerase chain reaction assay, across the proximal promoter region of the IL-2 gene. J Immunol 167, 4494–4503. PMID: 11591776