
The ultimate goal of a 2-D experiment is often the identification of differentially expressed proteins. This is achieved by mass spectrometric analysis of individual protein spots identified on a 2-D gel. The spots of interest are excised from the gel and the protein is subjected to proteolytic treatment prior to identification by mass spectrometry. This section reviews the spot excision methods and protein identification tools available.
Related Topics: Protein Sample Preparation for 2-D Electrophoresis, First Dimension Separation (Isoelectric Focusing), Second Dimension Separation, Staining and Visualization of Proteins After 2-D Electrophoresis, Imaging and Analysis of 2-D Electrophoresis Gels, and Troubleshooting 2-D Electrophoresis Gels with 2-D Doctor™.
Page Contents
Differentially expressed proteins identified by image analysis software such as PDQuest™ software can be marked on a printed image of the protein gel (see figure below), and the spots can then be manually cut from the gel.
There are two important factors to consider when handling 2-D gels for imaging and spot picking:
- Gels must be handled with gloves and placed on clean surfaces free of contamination. Keratin contamination from skin and hair is the leading cause of inconclusive mass spectrometry results. Use of hair nets, gloves, and masks is common practice when handling protein gels
- Gels must be cut in a clean, HEPA-filtered fume hood or via an automated spot cutter with a contained environment. Additional caution must be taken when manually cutting spots from fluorescent-stained 2-D gels using a UV transilluminator to ensure that skin and eyes are protected during the excision process, which can take anywhere from a few minutes to several hours, depending on the number of spots excised
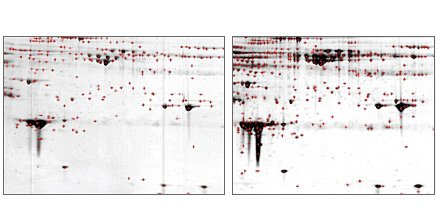
Protein spot identification with PDQuest software. Comparison of Coomassie (left panel) versus Flamingo™ (right panel) stains on ProteoMiner™ kit-treated serum samples on 2-D gel. Spot identification was performed using PDQuest software with identical spot identification parameters. Red crosses denote identified spots. A human serum sample was enriched in low-abundance proteins using the ProteoMiner protein enrichment kit. There were 249 and 402 spots detected in the Coomassie and Flamingo stained gels, respectively.
Identification of proteins is typically accomplished by in-gel digestion of protein spots followed by mass spectrometric analysis of extracted peptides. Excised gel plugs are destained and enzymatically digested (usually with trypsin). Protein identification can be accomplished using a variety of approaches including peptide mass fingerprinting using matrix-assisted laser desorption ionization with time of flight mass spectrometry (MALDI — TOF MS), MALDI tandem MS using MALDI—TOF—TOF mass spectrometry, or by liquid chromatography tandem (LC) MS using reversed-phase chromatography coupled online to the mass spectrometer via an electrospray ionization source. In the first approach, proteins are identified by comparing the peptide masses against a protein sequence database. In the latter two approaches, proteins are identified using peptide masses and their MS-MS fragmentation patterns to search the protein database.
For many proteomics projects, the identification of the proteins on 2-D gels is only the beginning of the research project. ExPASy proteomics tools (http://ca.expasy.org/tools) is a portal to the most popular web-based tools for in silico characterization of proteins based on their primary sequences. Taking time to filter a list of proteins to find those that make sense within the study parameters is worthwhile, because the next steps involve the biochemical characterizations of the short-listed proteins, which can be time consuming and costly. The functional characterization of proteins requires multiple approaches, including protein expression, purification, and production of antibodies. With these tools, many biochemical techniques can be used to further characterize proteins and their interacting partners (Figeys 2004).

