
Real-time PCR relies on highly specific and efficient amplification of the target sequence and the inclusion of fluorescent reporter molecules that enable the quantification of PCR products in real time. The use of a PCR primer specifically designed and validated for qPCR assays with your target of interest is highly recommended (see also qPCR Assay Design and Optimization). Fluorescent reporter molecules can be DNA-binding dyes such as SYBR® Green or fluorescently labeled PCR primers or probes. There are many different kinds of fluorescent dyes, PCR primers, and probes available for different applications. This section describes the various types of fluorescent dyes, PCR primers, and probes, and their specific uses in real-time PCR experiments.
Related Topics: qPCR Assay Design and Optimization and High Resolution Melting.
Page Contents
The most commonly used DNA-binding dye for real-time PCR is SYBR® Green, which binds nonspecifically to double-stranded DNA (dsDNA). SYBR® Green exhibits little fluorescence when it is free in solution, but its fluorescence increases up to 1,000-fold when it binds to dsDNA (Figure 1). Therefore, the overall fluorescence signal from a reaction is proportional to the amount of dsDNA present and increases as the target is amplified and more PCR product accumulates. As target amplification proceeds exponentially until the DNA primers are depleted, the fluorescence signal also increases exponentially, forming the basis for calculation of the original template quantity by the extrapolation of the real-time amplification curves.
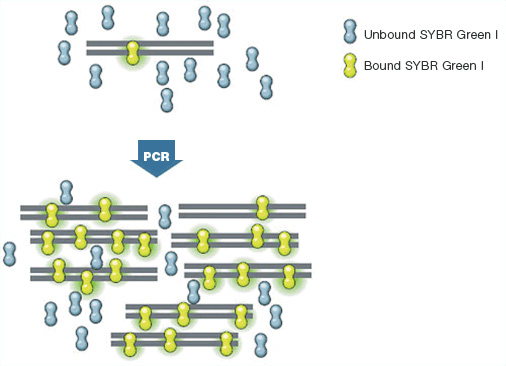
Fig. 1. SYBR® Green fluorescence dramatically increases when the dye molecules bind to dsDNA.
DNA-binding dyes can be classified as either nonsaturating or saturating. Saturating dyes — for example, SYBR® Green — inhibit PCR reactions when used above a certain (saturation) concentration. Nonsaturating dyes — for example, EvaGreen — do not inhibit PCR and can be used at a sufficiently high concentration to ensure that all vacant binding sites on dsDNA are bound by dye. However, saturating dyes are preferred for use in high resolution melt assays due to the more discrete signal change observed upon DNA denaturation (see High Resolution Melting (HRM)).
The advantages of using dsDNA-binding dyes include simple PCR primer design (only two sequence-specific DNA primers are needed, so probe design is not necessary), the ability to test multiple genes quickly without the need for multiple probes (for example, in the validation of gene expression data from many genes in a microarray experiment), lower initial cost than probes, and the ability to perform a melt curve analysis to check the specificity of the amplification reaction.
The major drawback of DNA-binding dyes is their lack of specificity; DNA-binding dyes bind to all dsDNA. As a result, the presence of nonspecific products in a real-time PCR reaction, such as PCR primer-dimers, contributes to the overall fluorescence and reduces the accuracy of quantification. Furthermore, DNA-binding dyes cannot be used for multiplex reactions because fluorescence signals from different products cannot be distinguished when, for example, amplifying a control gene along with a target gene using different PCR primers in the same reaction mixture. Thus, to examine multiple genes in real-time PCR assays using SYBR® Green, it is necessary to set up parallel reaction mixtures with different PCR primer pairs in separate tubes, which can present a source of error in quantification.
Many fluorescent PCR primer- and probe-based chemistries have been devised and are available from different commercial vendors, including:
- Hydrolysis (TaqMan) probes
- Molecular beacons
- Dual hybridization probes
- Eclipse probes
- Amplifluor assays
- Scorpions PCR primers
- LUX PCR primers
- QZyme PCR primers
PCR primer- and probe-based detection chemistries share some common features. In general, these chemistries use some form of fluorescence quenching to ensure that target-specific fluorescence is detected only when amplicon from the product of interest is present. The PCR primer or target-specific oligonucleotide probe is labeled with a reporter fluorophore, and in most cases, its fluorescence is quenched when the specific target DNA sequence is not present. Usually, this quenching is accomplished by covalently attaching a quencher molecule to the DNA primer or probe in combination with some mechanism by which the reporter and quencher are separated when the primer or probe binds to its specific target sequence.
In real-time PCR, fluorescent DNA primers and probes offer two main advantages over DNA-binding dyes. First, these molecules specifically detect the target sequence, so nonspecific products are not detected and do not affect the accuracy of quantification. Second, the use of fluorescent DNA primers and probes enables single-tube multiplexing of qPCR reactions for multiple target sequences.
Hydrolysis (TaqMan) Probes
Hydrolysis assays (TaqMan or 5' nuclease assays) include a sequence-specific, fluorescently labeled oligonucleotide probe in addition to a sequence-specific PCR primer. Hydrolysis assays exploit the 5' → 3' exonuclease activity of certain thermostable polymerases such as Taq or Tth. The hydrolysis probe is labeled with a fluorescent reporter at the 5' end and a quencher at the 3' end. Commonly used fluorescent reporter-quencher pairs are fluorescein (FAM), which emits green fluorescence, and Black Hole Quencher 1 dye.
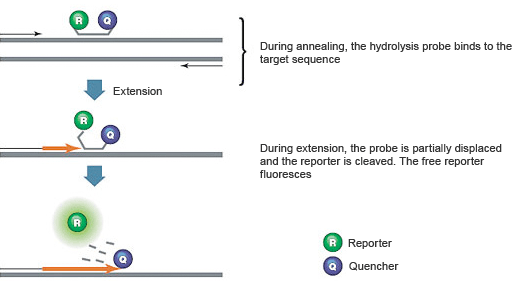
Fig. 2. Hydrolysis probes are hydrolyzed by Taq polymerase.
When the hydrolysis probe is intact, the fluorescence of the reporter is quenched due to its proximity to the quencher (Figure 2). The amplification reaction includes a combined annealing/extension step during which the probe hybridizes to the target, and the dsDNA-specific 5' → 3' exonuclease activity of Taq or Tth cleaves off the reporter. As a result, the reporter is separated from the quencher, resulting in a fluorescence signal that is proportional to the amount of amplified product in the sample.
The main advantages of using hydrolysis probes are high specificity, a high signal-to-noise ratio, and the ability to perform multiplex reactions. The disadvantages are that the initial cost of the probe may be high, and the assay design may not be trivial.
Molecular Beacons
Molecular beacon assays include a sequence-specific, fluorescently labeled oligonucleotide probe called a molecular beacon. A molecular beacon is a dye-labeled oligonucleotide (25–40 nt) that forms a hairpin structure with a stem and a loop (Figure 3). The 5' and 3' ends of the probe have complementary sequences of 5–6 nucleotides that form the stem structure. The loop portion of the hairpin is designed to specifically hybridize to a 15–30 nucleotide section of the target sequence. A fluorescent reporter molecule is attached to the 5' end of the molecular beacon, and a quencher is attached to the 3' end. Formation of the hairpin therefore brings the reporter and quencher together, so no fluorescence is emitted.
![]()
Fig. 3. The stem-loop structure of molecular beacons brings the fluorophore and quencher together.
During the annealing step of the amplification reaction, the loop portion of the molecular beacon binds to its target sequence, causing the stem to denature. The reporter and quencher are thus separated, quenching is abolished, and the reporter fluorescence is detectable. Because fluorescence is emitted from the probe only when it is bound to the target, the amount of fluorescence detected is proportional to the amount of target in the reaction.
Molecular beacons have some advantages over other probe chemistries. They are highly specific, can be used for multiplexing, and if the target sequence does not match the beacon sequence exactly, hybridization and fluorescence will not occur, which is especially desirable for allelic discrimination experiments. Unlike hydrolysis assays, molecular beacons are displaced but not destroyed during amplification because a DNA polymerase lacking 5' exonuclease activity is used. The main disadvantage of using molecular beacons is that they are difficult to design. The stem of the hairpin must be strong enough that the molecule will not spontaneously fold into nonhairpin conformations that result in unintended fluorescence. Conversely, the stem of the hairpin must not be too strong, or the beacon may not properly hybridize to the target.
Dual Hybridization Probes
These assays use two sequence-specific oligonucleotide probes in addition to two sequence-specific DNA primers. The two probes are designed to bind to adjacent sequences in the target (Figure 4). The probes are labeled with a pair of dyes that exhibit fluorescence resonance energy transfer (FRET). The donor dye is attached to the 3' end of the first probe, while the acceptor dye is attached to the 5' end of the second probe.
During real-time PCR, excitation is performed at a wavelength specific to the donor dye, and the reaction is monitored at the emission wavelength of the acceptor dye. At the annealing step, the probes hybridize to their target sequences in a head-to-tail arrangement. This annealing brings the donor and acceptor dyes into proximity, allowing FRET to occur, resulting in fluorescent emission by the acceptor. The increasing amount of acceptor fluorescence is proportional to the amount of PCR product present.
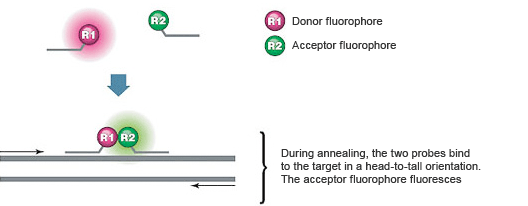
Fig. 4. FRET occurs when dual hybridization probes bind to their target sequences.
Eclipse Probes
These assays use two DNA primers and a sequence-specific oligonucleotide probe. The Eclipse probe (Figure 5) is complementary to a sequence within the target and has a fluorescent reporter molecule at the 3' end combined with a quencher and a DNA minor-groove binder at the 5' end. The unhybridized probe adopts a random coil conformation that brings the reporter and quencher together, so quenching occurs. During the annealing of the DNA primers, the probe also hybridizes to the target with the help of the minor-groove binder. The probe thus becomes linearized, separating the reporter and quencher. The resulting fluorescence signal is proportional to the amount of amplified product in the sample.
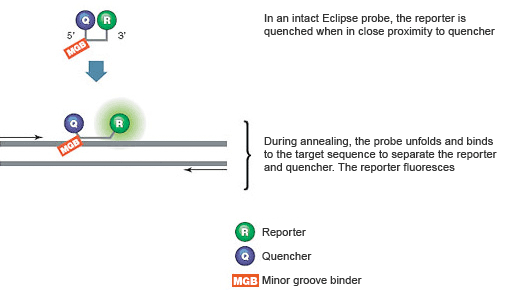
Fig. 5. The conformation of the unhybridized Eclipse probe causes quenching.
Amplifluor Assays
These assays use two target-specific DNA primers and one universal DNA primer called a UniPrimer. An 18-nucleotide sequence called the Z sequence extends from the 5' end of one target-specific PCR primer (called the Z primer); this same sequence forms the 3' end of the UniPrimer. The UniPrimer forms a hairpin structure, with a fluorescent reporter and a quencher attached to the 5' and 3' ends of the stem, respectively. In the hairpin conformation, the reporter and quencher are brought into proximity, so fluorescence is quenched. During the first amplification cycle, the Z primer hybridizes to the template and is extended (Figure 6). During the second amplification cycle, the other target-specific DNA primer hybridizes to the product of the first and serves as a template for the synthesis of a new target strand that contains a sequence complementary to the Z sequence. The product from the second amplification cycle can then serve as the template for the UniPrimer. In the third amplification cycle, the extended UniPrimer serves as a template for the next amplification cycle. In the fourth cycle, extension of the template through the UniPrimer causes the hairpin to open and adopt a linear configuration. The reporter and quencher are thus separated and fluorescence is emitted. Exponential amplification using the second target-specific DNA primer and the UniPrimer occurs in subsequent PCR cycles. The resulting fluorescence signal is proportional to the amount of amplified product in the sample.
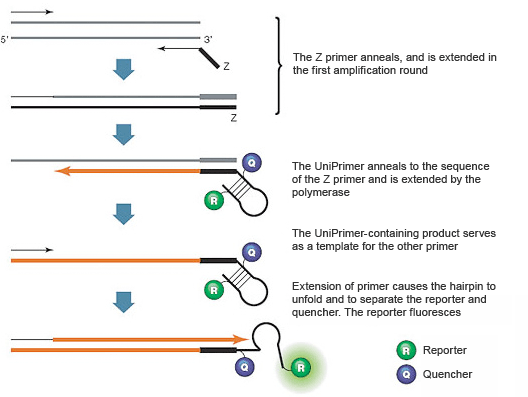
Fig. 6. Amplifluor assays use a special PCR primer called the UniPrimer.
Scorpions PCR Primers
These assays use two PCR primers, one of which serves as a probe and contains a stem-loop structure with a 5' fluorescent reporter and 3' quencher. The loop of the Scorpions probe includes a sequence that is complementary to an internal portion of the target sequence (Figure 7). During the first amplification cycle, the Scorpions PCR primer is extended, and the sequence complementary to the loop sequence is generated on the same strand. After subsequent denaturation and annealing, the loop of the Scorpions probe hybridizes to the internal target sequence, and the reporter is separated from the quencher. The resulting fluorescence signal is proportional to the amount of amplified product in the sample. The Scorpions probe contains a PCR blocker, just downstream (in the 3' direction) of the quencher, to prevent read-through during the extension of the opposite strand.
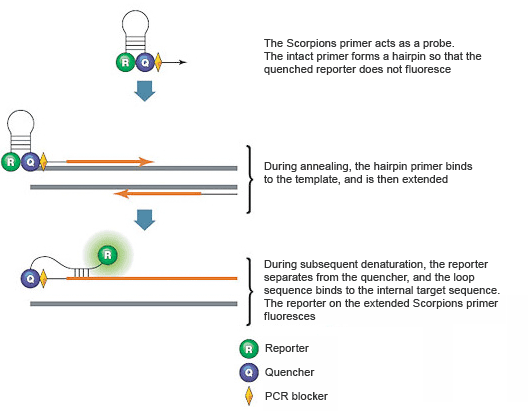
Fig. 7. Scorpions PCR primers contain a sequence complementary to an internal portion of the target sequence.
LUX PCR Primers
These assays employ two DNA primers, one of which is a hairpin-shaped PCR primer with a fluorescent reporter attached near the 3' end, as illustrated in Figure 8. The reporter is quenched by the secondary structure of the hairpin. During amplification, the LUX PCR primer is incorporated into the product, eliminating the quenching hairpin structure, so fluorescence is emitted.
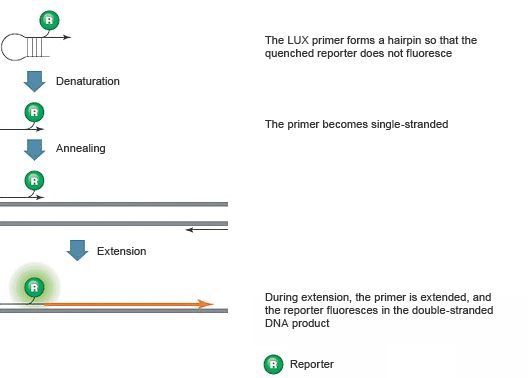
Fig. 8. The hairpin structure of unbound LUX PCR primers quenches the fluorophore.
QZyme PCR Primers
These assays use a target-specific PCR primer that contains an antisense (inactive) sequence for a catalytic DNA structure (a deoxyribozyme), a universal oligonucleotide substrate for the catalytic DNA, and a target-specific reverse primer, as shown in Figure 9. The oligonucleotide substrate contains a fluorescent reporter on the 5' end and a quencher on the 3' end. When the substrate is intact, the fluorescence of the reporter is quenched. During the first amplification cycle, the PCR primer containing the antisense catalytic sequence is extended. In the second cycle, the product of the first cycle serves as the template for the target-specific reverse primer, which is extended to create a new product that contains the sense (active) catalytic DNA region. In the subsequent annealing step, the fluorescently labeled oligonucleotide substrate hybridizes to the catalytic DNA sequence and is cleaved. This cleavage separates the reporter from the quencher, resulting in a fluorescence signal that is proportional to the amount of amplified product in the sample.
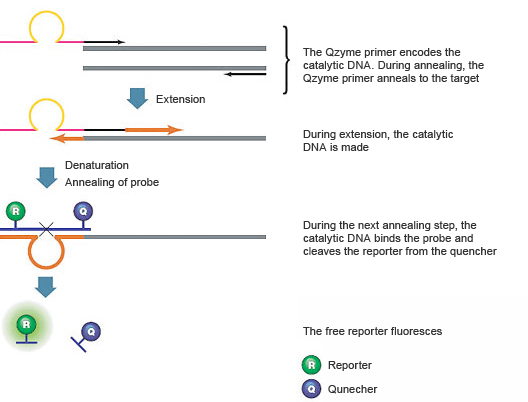
Fig. 9. QZyme assays use a catalytic DNA structure to cleave a labeled oligonucleotide substrate.
Gudnason H et al. (2007). Comparison of multiple DNA dyes for real-time PCR: Effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res 35, e127. PMID: 17897966
Wong ML and Medrano JF (2005). Real-time PCR for mRNA quantitation. BioTechniques 39, 75–85. PMID: 16060372